Page Contents
WHAT IS IT?
Hemophilia A is an X-linked recessive genetic condition that is marked by a deficiency in clotting factor VIII of the coagulation cascade. Mutations in the factor VIII gene are responsible for this condition (leading to decreased activity of the factor) and are usually inherited (although spontaneous mutations are also possible).
WHY IS IT A PROBLEM?
Factor VIII is an important clotting factor for the coagulation cascade. Its absence/deficiency predisposes the patient to bleeding. The severity of the condition is spectral given different levels of Factor VIII activity present in patients.
WHAT MAKES US SUSPECT IT?
Risk factors: male, family history
Common chief concerns: intuitively, many of the chief concerns patients with this condition will express revolve around episodes of bleeding.
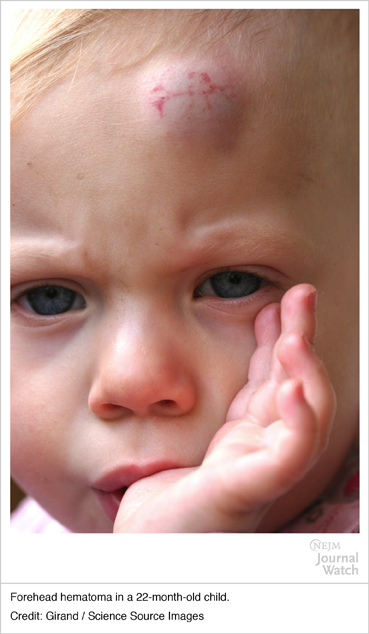
Hematoma (head): these bouts of bleeding are common in young children who frequently experience bumps to the head.
Easy/excessive bruising: such an observation should raise clinical suspicions for hemophilia
Prolonged bleeding after surgery: such an observation should raise suspicions for hemophilia (bleeding excessively after dental procedures is common)
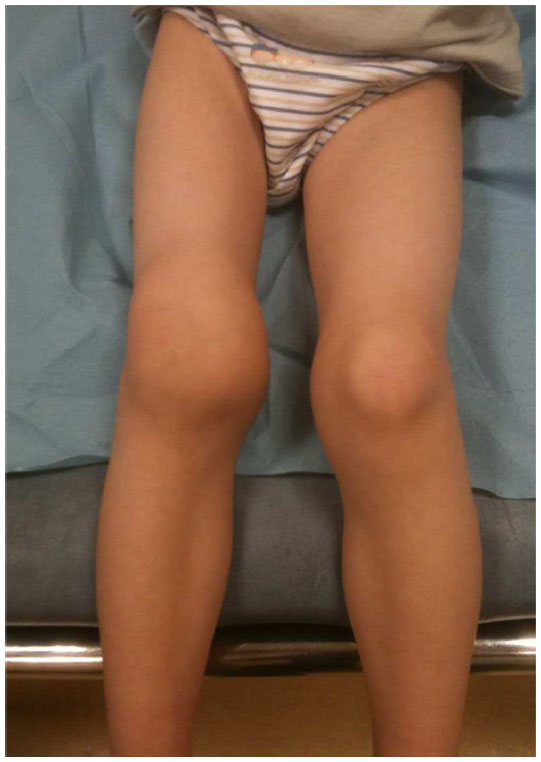
Hemarthroses: bleeding can occur in the joints (such as the knee) which can appear swollen on exam, and also can cause pain (and associated limping). Also called chronic hemophilic arthropathy, proteases form the blood in the synovial space will destroy joint cartilage.
HOW DO WE CONFIRM A DIAGNOSIS?
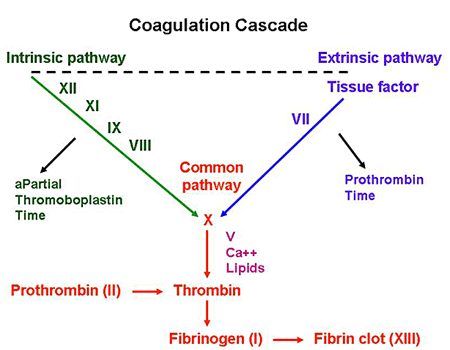
Coagulation studies: use this guide as a reference to interpret the below findings that one would see in hemophilia A.
- Increased PTT (factor VIII is in the intrinsic pathway)
- Normal PT
- Normal BT
Factor VIII activity assay: if the results of the coagulation factor are what is seen above, directly testing for factor VIII activity can assess the likelihood of hemophilia A. Factor VIII activity will be decreased in patients with this condition (and lower activities are associated with more severe disease). KEEP IN MIND THIS IS HOW WE CAN DISTINGUISH BETWEEN HEMOPHILIA A VS. B (they are initially clinically indistinguishable).
Genetic testing can be used to confirm the specific causal mutation for this condition.
HOW DO WE TREAT IT?
Desmopressin: this synthetic analog of the anti diuretic hormone ADH/vasopressin, works to help hemophiliacs in a number of ways:
- Increases release of both factor VIII and von Willebrand factor from endothelial cells (increase serum levels of these proteins).
- Stimulates platelet adhesion
- Increases expression of tissue factor (the inciting factor for the extrinsic arm of the coagulation cascade)
Factor VIII concentrate: recombinant factor VIII protein is given to the patients given that this is the clotting factor that they are missing.
HOW WELL DO THE PATIENTS DO?
This condition can be very well managed pharmacologically, and mortality of patients in resource rich parts of the world is comparable to that of the general population (source)
WAS THERE A WAY TO PREVENT IT?
While there is not way to prevent this genetic condition, screening potential female carriers of the disease can help inform patients on the risk of having children with hemophilia A.
WHAT ELSE ARE WE WORRIED ABOUT?
Intracranial hemorrhage while rare, this is the leading cause of bleeding-related death in patients with hemophilia A (source)
Patients can develop inhibitors (IgG antibodies that neutralize the clotting factor). Keep this in mind as it changes the treatment landscape for the patient! If a patient has inhibitors replacing the clotting factor will not be as beneficial.
OTHER HY FACTS?
If patients do not improve after recombinant factor VIII supplementation, test them for the presence of factor VIII inhibitors (antibodies that antagonize this factor which can develop over time in patients).
Symptomatic forms generally have less that 25% activity of factor VIII
Factor VIII is NOT vitamin K dependent
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
Page Updated: 01.09.2016