Page Contents
WHAT IS IT?
Sickle cell disease/sickle cell anemia is a homozygous recessive genetic disorder that is caused by the production of hemoglobin with an altered amino acid sequence, caused by a single point mutation in the β-globin gene.
WHY IS IT A PROBLEM?
The causal point mutation results in a protrusion on the ß-globin chains that create a distinct type of hemoglobin (called HbS). In low oxygen environments HbS will polymerize into longer fibers that will cause sickling of red blood cells (RBCs), which ultimately occlude blood flow in the capillaries. These occlusions will cause micro-infarcts in tissues causing severe hypoxia and pain. Furthermore, the spleen is one of the first affected organ because it’s environment is the most conducive to facilitating the sickling of RBC’s (large complicated vasculature where RBCs can aggregate).
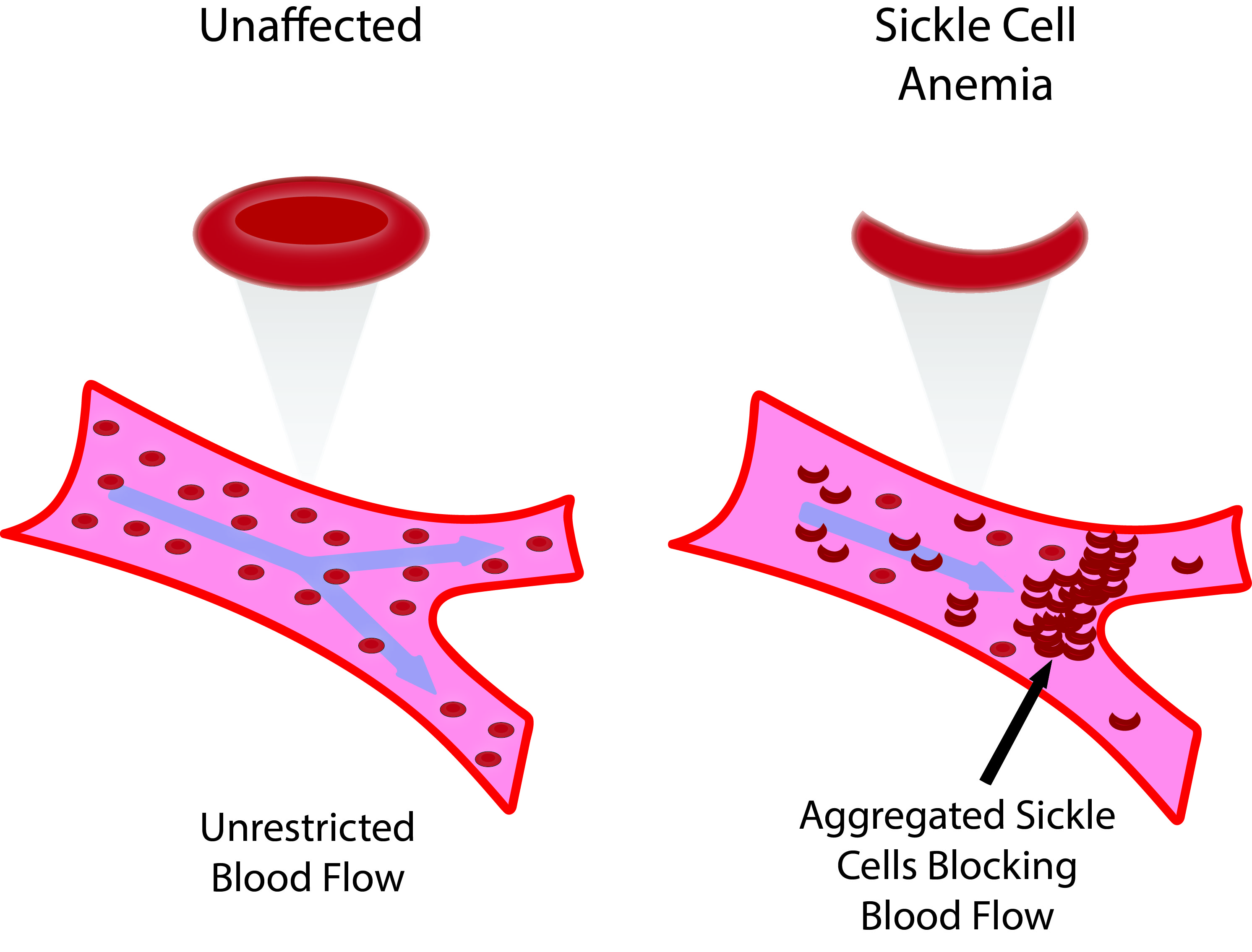
Sickled red blood cells are more fragile, so normal mechanical stresses experienced by RBCs in the systemic circulation will result in hemolysis. Patients will have a chronic hemolytic anemia as a result.
WHAT MAKES US SUSPECT IT?
Risk factors: family history, african american race,
Pain: acute vascular occlusions will cause periodic episode of pain.
Symptoms of Organ disfunction: occlusion of vessels can manifest in a wide variety of issues that can include pain in the bones, chest pain, shortness of breath, leg ulcers, stroke (and associated symptoms), renal papillary necrosis, and priapism.
Splenomegaly: Trapping of sickled RBCs in the spleen can cause splenomegaly. This will be a finding usually only in younger patients, because after time, the spleen will cirrhoses and atrophy (in progressed disease it will no longer be palpable).
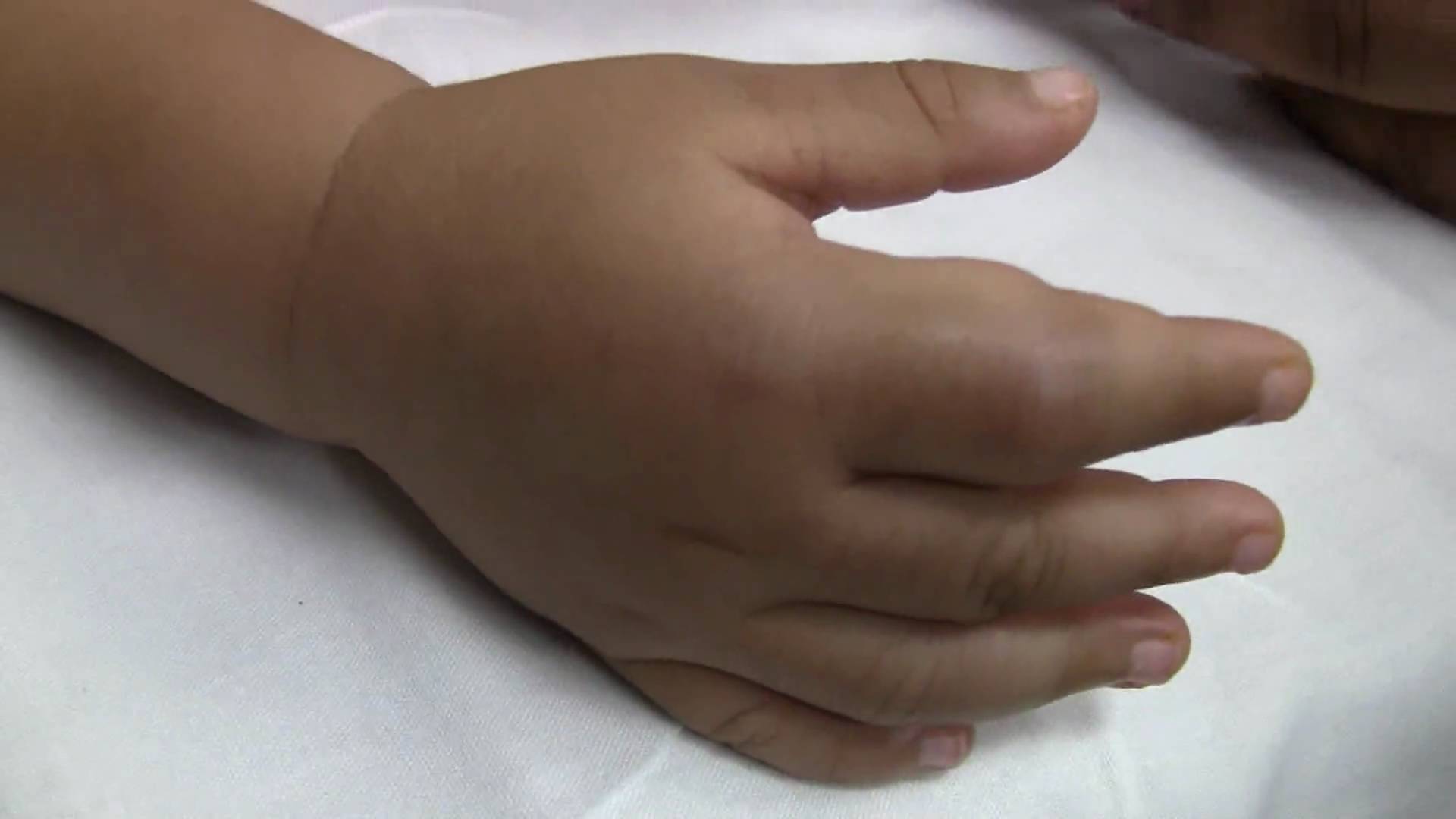
Dactylitis (hand-foot syndrome): swelling of the hands or feet usually seen in infants.
HOW DO WE CONFIRM A DIAGNOSIS?
Complete blood count (CBC): Hemoglobin (Hb) and hematocrit (Hct) are decreased (mean corpuscular volume normal). Reticulocyte count is increased. All of these findings are consistent with hemolytic anemia.
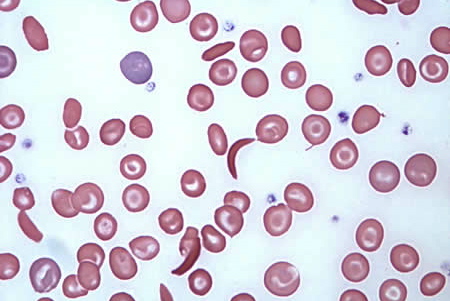
Blood smear: A peripheral blood smear will reveal the following findings:
Crescent shaped RBCs: these are the sickled cells discussed above.
Target cells (Codocytes): relative membrane excess found on RBCs leading to this pathological finding.
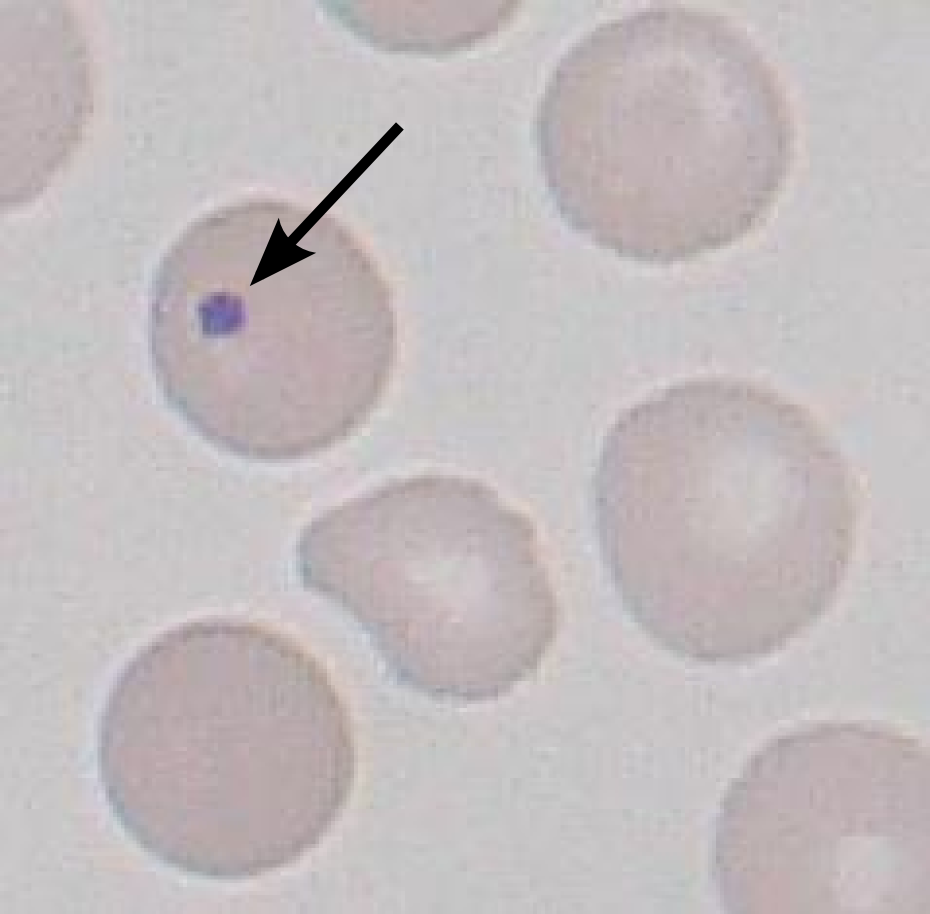
Howell Jolly Bodies: small portions of remnant DNA found in RBCs (which normally lose their entire nucleus upon development). The presence of these Howell Jolly bodies suggests the absence of either the spleen, or issues with its proper function. Sickle cell patients usually will have impaired spleen function.
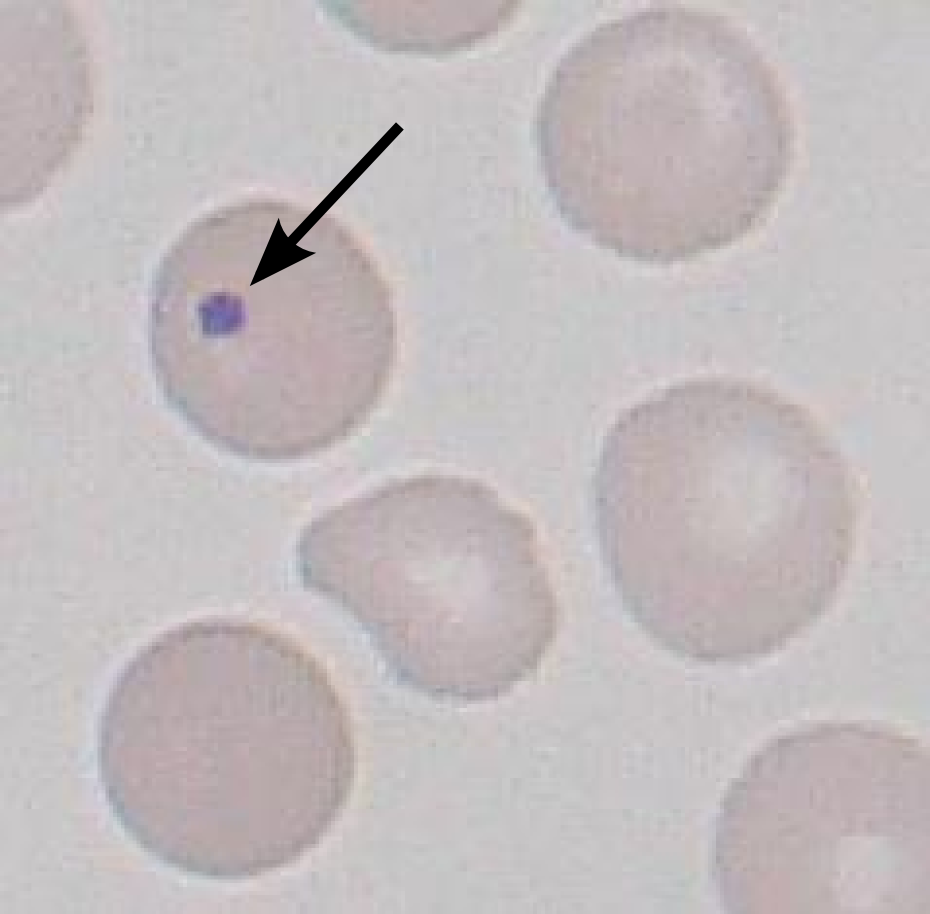{kind=link}
Sickle cell screen: the addition of sodium metabisulfite (which reduces the present oxygen in the sample) will induce sickling of cells and this can be used as a means to screen for the disease quickly.
Hemoglobin electrophoresis: this will reveal the absence of hemoglobin A (HbA), increased HbS, and often increased levels of fetal hemoglobin (HbF) as well. Carriers will have both HbA and HbS.
HOW DO WE TREAT IT?
Hydroxyurea: is used to treat sickle cell disease because it increases circulating levels of HbF, which ultimately decreases RBC sickling (HbF stops the aggregation of HbS) .
Oxygen therapy: this can stop early sickling.
Antibiotics: Loss of splenic function makes patients increasingly susceptible to infection by encapsulated bacteria and daily antibiotic prophylaxis is required for the treatment of children.
Immunizations: patients with sickle cell disease must be up to date on their immunizations (pneumococcal, meningococcal vaccine, haemophilus influenzae type B, hepatitis B, and influenza vaccines).
Pain medications: such as morphine can assist with pain relief.
Folic acid supplementation: all patients with sickle cell anemia should receive folic acid to assist with RBC development.
Exchange transfusion: patients sufferenign from a suspected stroke (secondary to RBC sickling) are treated acutely with an exchange transfusion.
HOW WELL DO THE PATIENTS DO?
Patients with sickle cell disease report having pain on most days even with proper management (source).
WAS THERE A WAY TO PREVENT IT?
Given its genetic cause, there is no way to prevent sickle cell anemia. With this in mind, all newborns are screened for sickle cell anemia.
WHAT ELSE ARE WE WORRIED ABOUT?
Acute Chest Syndrome: this is a complication of vascular occlusion that presents similarly to pneumonia (and can be precipitated by pneumonia). Symptoms including fever, cough, pain, production of sputum, dyspnea, or low oxygen levels. Chest X-ray can demonstrate pulmonary infiltrates. Treatment is with broad spectrum antibiotics (for possible infection), pain control, supplemental O2, and transfusion.
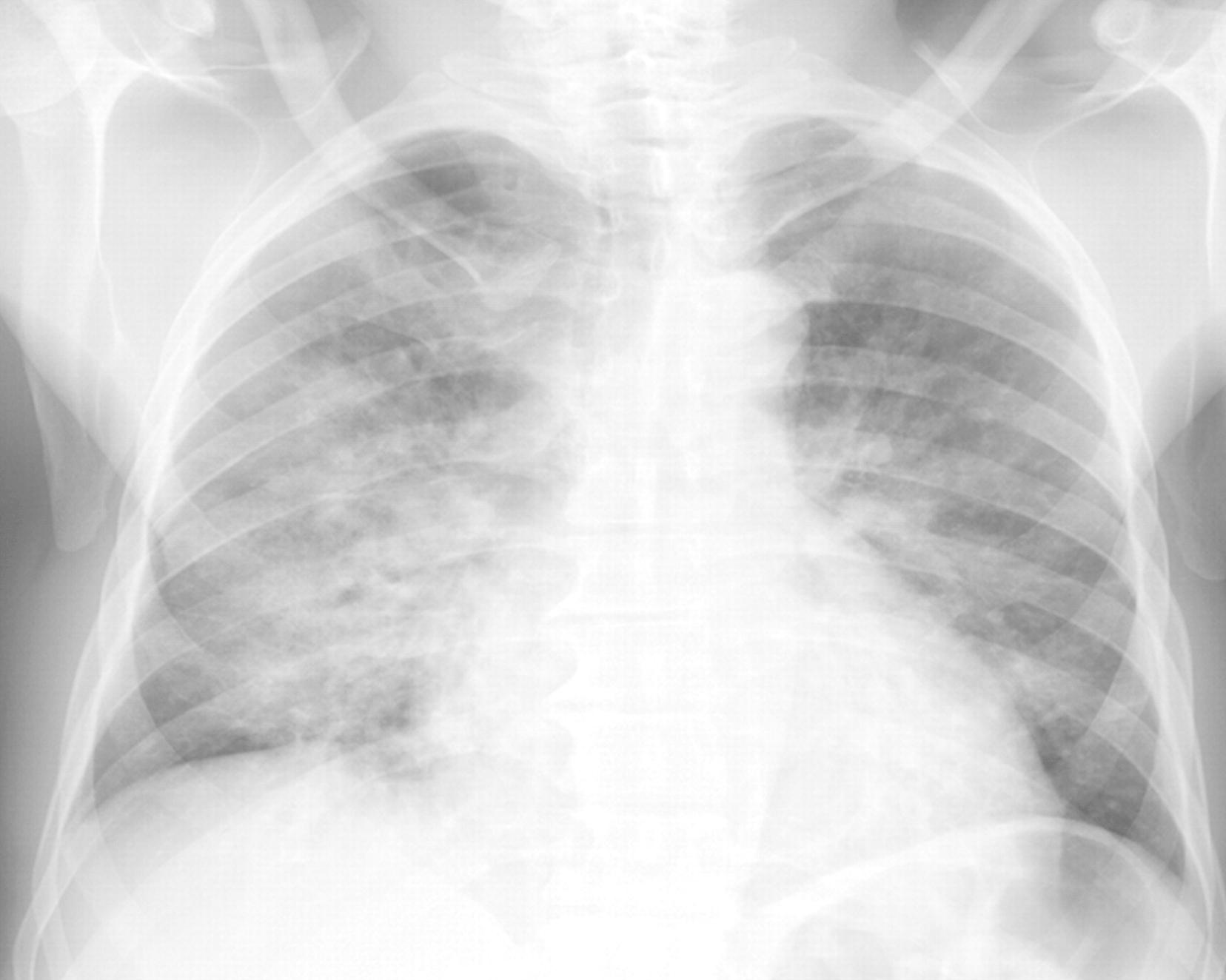
Stroke: patients with sickle cell disease are at higher risk for stroke. While occlusions are not caused necessarily by the accumulation of sickling cells, these patients have a proliferative vasculopathy of arteries leading to the brain, which predisposes/sensitizes them to stroke.
Infections: Patients with sickle cell will undergo autosplenectomy (excessive damage to the spleen that renders it nonfunctional) at a young age. Children with impaired spleen function are at higher risk for infection (pneumococcal sepsis used to be a large cause of death in patients). Prophylactic penicillin used for newborns, and children given the pneumococcal vaccine. Osteomyelitis is associated with salmonella infection in patients with sickle cell.
Pain crisis: as explained above, micro infarcts in tissues will result in episodes of pain that can be difficult to manage. Make sure to keep in mind this element of the disease, as there will be limited physical exam finings to support pain in many of the region patients are experiencing them (don’t assume patients are “faking” pain!).
OTHER HY FACTS?
Causal mutation: the mutation is a glutamate-to-valine mutation at position 6 in the β-globin chain.
Aplastic crisis us association with parvovirus B19. This is a stop in the production of RBCs (decreased reticulocytes seen here).
Erythrocyte sedimentation rate (ESR) decreased by sickle cell.
Causes of sickling: the process of sickling is increased by anything that raises the proportion of HbS in the deoxygenated state. This includes:
- decreased O2 availability,
- increased CO2 in the blood
- decreased pH
- dehydration
- increased 2,3-BPG
Page Updated: 01.19.2016