Page Contents
WHAT IS IT?
McArdle disease is an autosomal recessive, type V glycogen storage disease. It is the most common glycolytic disorder.
WHAT CAUSES IT?
A deficiency in the skeletal muscle glycogen phosphorylase (myophosphorylase) is ultimately the cause of this condition. This condition can be traced back to a mutation on the PYGM (phosphorylase, glycogen, muscle) gene, located on chromosome 11q13.1. There have been over 140 pathogenic mutations identified on this been (mis-sense/nonsense mutations being the most common).
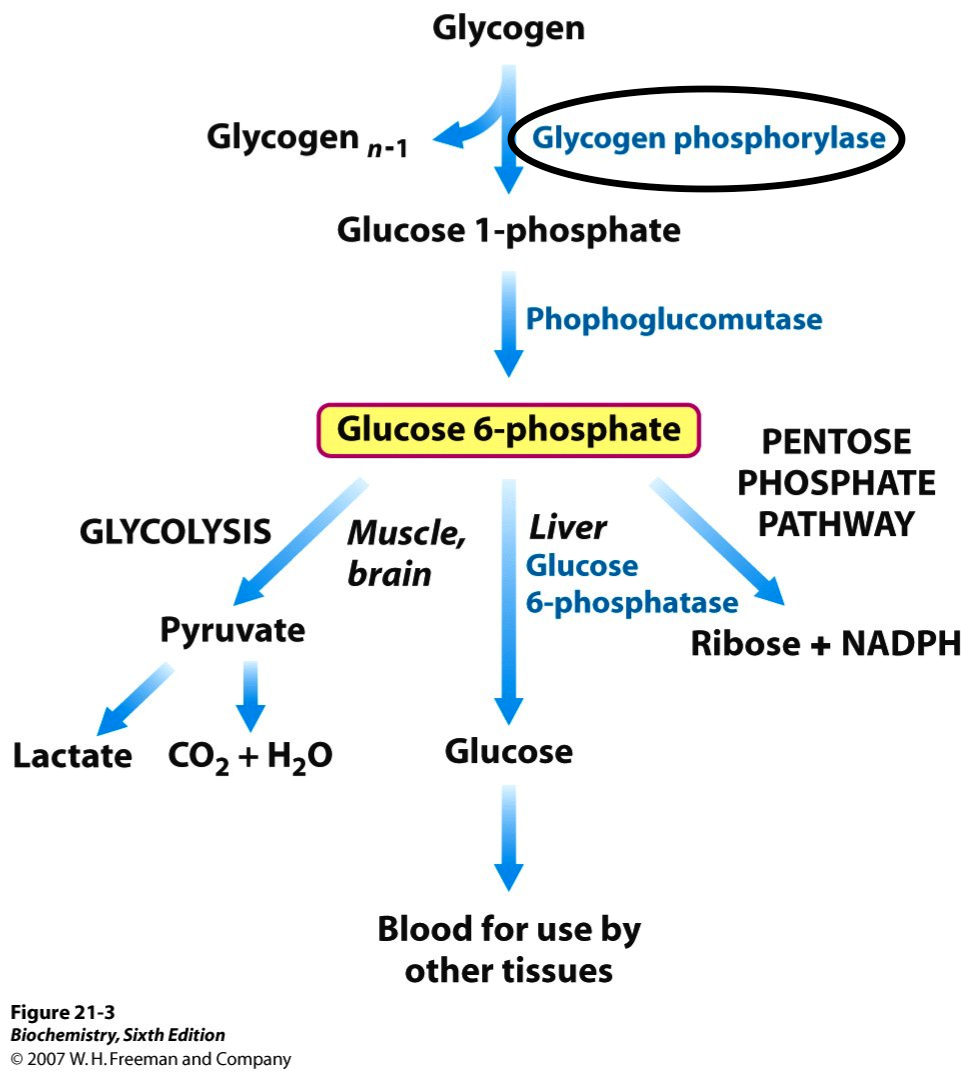
WHY IS IT A PROBLEM?
In the absence of myophosphorylase activity, the glycogen stored in muscles cannot be broken down and used to generate ATP in muscle tissues. During strenuous exercise this will force muscle tissues to rely upon free fatty acid metabolism, and will result in painful muscle cramps. Whats more, this condition can even result in rhabdomyolysis as muscle tissues are damaged.

WHAT MAKES US SUSPECT IT?
Risk factors
Family history of McArdle disease
Initial Presentation
Common Chief Complaints:
- Exercise intolerance that is characterized by fatigue, painful cramps, and myalgia
- Red/dark urine due to myoglobinuria
History Of Present Illness
Onset: patients will notice painful muscle cramps during exercise, and only after these periods of exercise will they notice dark/red urine.
Symptoms are most noticeable with brief, intense bouts of exercise.
Second wind phenomenon: during the exercise, patients may experience a “second-wind” due to a period of increased muscle blood flow/mobilization of blood glucose. This often occurs after ~10 minutes of painful exercise.
Physical Exam Findings
Generally speaking the physical exam can be quite unremarkable in patients (due to the nature of this condition).
In certain settings there may patterns of muscle weakness (due to repeat muscle damage)
CLINICAL WORKUP
Blood glucose: while a little unexpected, patients with condition will generally not have abnormally low levels of blood glucose.
Serum creatine kinase levels will often be increased in patients with this condition (due to muscle damage).
Urinalysis can reveal the presence of myoglobin in the urine (in the absence of red blood cells).
Forearm nonischemic test: patients perform hand grip exercises repetitively and then will have their serum lactate levels measured.
- In unaffected levels the level of serum lactate will increase as glucose is utilized by lactic acid fermentation.
- In affected patients serum lactate levels will NOT increase given that the glycogen in the muscles can not be broken down.
Genetic analysis of the PYGM can reveal the presence of the casual mutation in patients (this ultimately confirms the diagnosis)
Muscle biopsy can be useful in patient’s with inconclusive:
- Biochemical analysis of muscle myophosphorylase enzyme activity will be decreased in affected patients.
- Histological staining of biopsied tissues will demonstrate decreased myophosphorylase activity and increased presence of glycogen.
HOW DO WE TREAT IT?
Sucrose supplementation before beginning exercise can provide some benefit.
Engaging in moderate level exercise (aerobic activity) can be beneficial. Patients should avoid extreme short bouts of intense exercise to prevent muscle damage.
HOW WELL DO THE PATIENTS DO?
This glycogen storage disorder is comparatively mild in nature, and patients (in the absence of serious complications such as renal failure) will have a favorable prognosis.
WAS THERE A WAY TO PREVENT IT?
This genetic condition is not preventible, however rhabdomyolysis can be avoided by limiting the nature of the exercise (moderate, aerobic exercise should not lead to muscle damage).
WHAT ELSE ARE WE WORRIED ABOUT?
Cardiac arrhythmia can corm due to electrolyte abnormalities in this condition.
Renal failure is a concern with prolonged exercise. This is a result of damage to the kidney due to myoglobinuria.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 08.01.2016