Page Contents
WHAT IS IT?
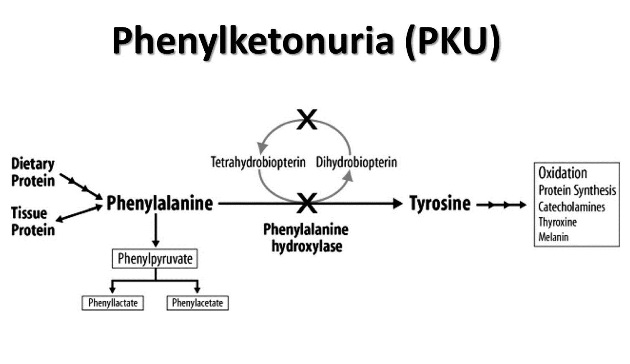
Phenylketonuria (PKU) is an autosomal recessive disease that is typically caused by a defeincy of phenylalanine hydroxylase (PAH). It can also be caused by a deficiency of tetrahydrobiopterin cofactor/BH4 (this condition is specifically referred to as malignant PKU). Fundamentally, is a disease of amino acid metabolism (phenylalanine).
WHY IS IT A PROBLEM?
The lack of phenylalanine hydroxylase activity will lead to increased phenylalanine in the body (and subsequent increased phenylketones in the urine). This increase in phenylalanine will cause a variety of issues in the patient including intellectual disability and growth retardation. This increase in phenylalanine will also lead to elevated levels of phenylalanine metabolites such as phenyllactate, phenylacetate, and phenylpyruvate, which give urine a characteristic “musty” or “mousey” odor.
Tyrosine will also be deficient in patients because it is derived from phenylalanine.
WHAT MAKES US SUSPECT IT?
Risk factors:
Clinical presentation can include light skin, intellectual disability, developmental delay (growth retardation), and musty body odor.
Other signs can include seizures, microcephaly,tremor and eczema.
Hypopigmentation is common in patients (fair her, blue eyes, light skin color) simply because melanin formation starts with hydroxylation of tyrosine, a process inhibited by high levels of phenylalanine in PKU.
HOW DO WE CONFIRM A DIAGNOSIS?
Serum analysis of phenylalanine is the initial step in screening patients for PKU. Levels above > 120 mcmol/L are suspicious.
Genetic testing can reveal the causal mutation either on the PAH gene or BH4 gene.
HOW DO WE TREAT IT?
Phenylalanine restriction in the patient’s diet.
- Aspartame (artificial sweetener) must be avoided in PKU patients because it contains phenylalanine.
Tyrosine increase in the patient’s diet.
Tetrahjydrobiopterin supplementation is needed for those with malignant PKU.
HOW WELL DO THE PATIENTS DO?
Patients who are diagnosed with PKU at birth have an excellent prognosis (and will have no long term complications).
WAS THERE A WAY TO PREVENT IT?
This genetic condition is not preventible, but is screened for in the US
Newborn screening for this condition occurs 2-3 days after birth because at birth enzyme levels will be normal due to maternal supply in the baby’s blood stream.
WHAT ELSE ARE WE WORRIED ABOUT?
Missing the diagnosis in newborns and the development of preventible intellectual disability (evident at 1 year of age).
OTHER HY FACTS?
This condition is an example of pleiotropy
Phenylketone examples include: phenylacetate, phenyllactate, and phenylpyruvate.
Maternal PKU refers to a lack of proper dietary therapy during pregnancy. Infants will have microcephaly, intellectual disability, growth retardation, and congenital heart defects.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 06.13.2016