Page Contents
WHAT IS IT?
Acute myocardial infarction (acute MI) refers to what is commonly called a “heart attack” and belongs to the broader category of diseases that are refereed to as ischemic heart disease. During an MI, the myocardium of the heart is ischemic for so long that there is some degree of irreversible damage due to the necrosis of myocardial cells.
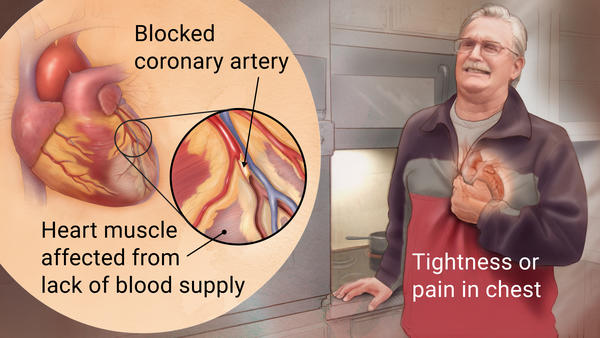
Most often this is caused by a thrombosis due to the rupture of a coronary artery atherosclerotic plaque however other disease processes can cause an MI:
- Systemic amyloidosis
- Coronary artery vasospasm (due to Prinzmetal angina or cocaine use)
- Vasculitis (such as Kawasaki disease)
- Emboli
- Usage of selective COX-2 inhibitors (lead to prothrombogenic states)
- Hypertensive emergency
- Hyper viscosity syndrome
It is important to appreciate that there are different types of MIs.
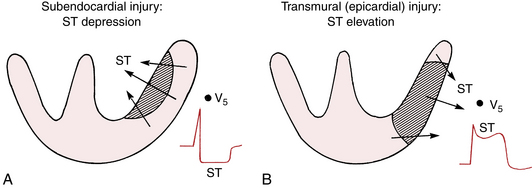
Subendocardial infarction occurs in the initial phases of an MI. It involves less then 50% of the myocardial wall thickness (usually only the innermost layer of the heart which is the furthest from the blood supply). Continued ischemia can lead to transmural infarction.
Transmural infarction involves most (if not all) of the myocardial wall (usually all three layers)
WHY IS IT A PROBLEM?
Given an MI is defined by the irreversible damage of the myocardium, the ability of the heart to pump blood is compromised by this disease process. The cardiac wall often is dysfunctional post MI.
WHAT MAKES US SUSPECT IT?
Risk factors: atherosclerosis (CAD), hyperglycemia (diabetes), homocystinuria, familial hypercholesterolemia (type IIa), Triptans usage (higher risk of coronary spasm)
Chief complaints can include:
- Chest pain that is severe and crushing in nature. Radiates to the left arm/jaw. NOT releived by administration of nitrates like nitroglycerin.
- Diaphoresis
- Dyspnea
- Nausea and vomiting
- Fatigue
Pain is not usually associated with:
- Pleuritic chest pain that gets worse with inspiration/coughing
- Provocation with physically touching the chest
- Very short (few seconds) of pain.
- Radiation of pain to the lower extremities.
- Localization of pain with 1 fingertip
HOW DO WE CONFIRM A DIAGNOSIS?
EKG is very useful in helping diagnose an MI. There can be different types of findings:
- ST Depressions can be seen with with an NSTEMI and are typically associated with subendocardial MIs.
- T wave inversions are also associated with subendocardial MIs.
- ST elevations are diagnostic of a STEMI and typically are associated with transmural MIs
- Pathologic Q waves can be seen with transmural MIs.
Elevated cardiac biomarkers are diagnosis of an MI.
- Troponin I is the most sensitive and specific marker (gold standard). Stays elevated for ~ a week.
- CK-MB can detect reinfarction that occurs days after initial MI (it returns to normal ~72 hours after initial infarction).
HOW DO WE RULE OTHER DIAGNOSES OUT?
The EKG and ultimately the cardiac biomarker lab tests can help identify an MI given the broad differential diagnosis that can surround the presentation of acute chest pain.
HOW DO WE TREAT IT?
Management of MI is complicated and depends on a few factors (i.e. STEMI vs. NSTEMI). The below is a generalization.
Immediate treatment involves:
- Aspirin as soon as possible to limit thrombosis
- Nitroglycerin to reduce preload (can be taken by patient suspecting ischemia prior to the hospital visit)
- Chair/bed rest with continuous EKG monitoring.
- Supplemental oxygen to minimize ischemia
- Morphine for pain, anxiety, and pulmonary edema
Within 24 hours of presentation:
- β-blocker to slow the hear rate, and decrease oxygen demand of the heart
- ACE-inhibitor to reduce afterload on the heart (especially for those with hypertension and reduced left ventricular ejection fraction
- Anti-platelet therapy (ex. clopidogrel) can be started as well
- Heparin to further prevent coagulation.
Invasive treatment strategies may be required in some patients. This involves cardiac catheterization and angioplasty in order to re-vascularize ischemic portions of the myocardium.
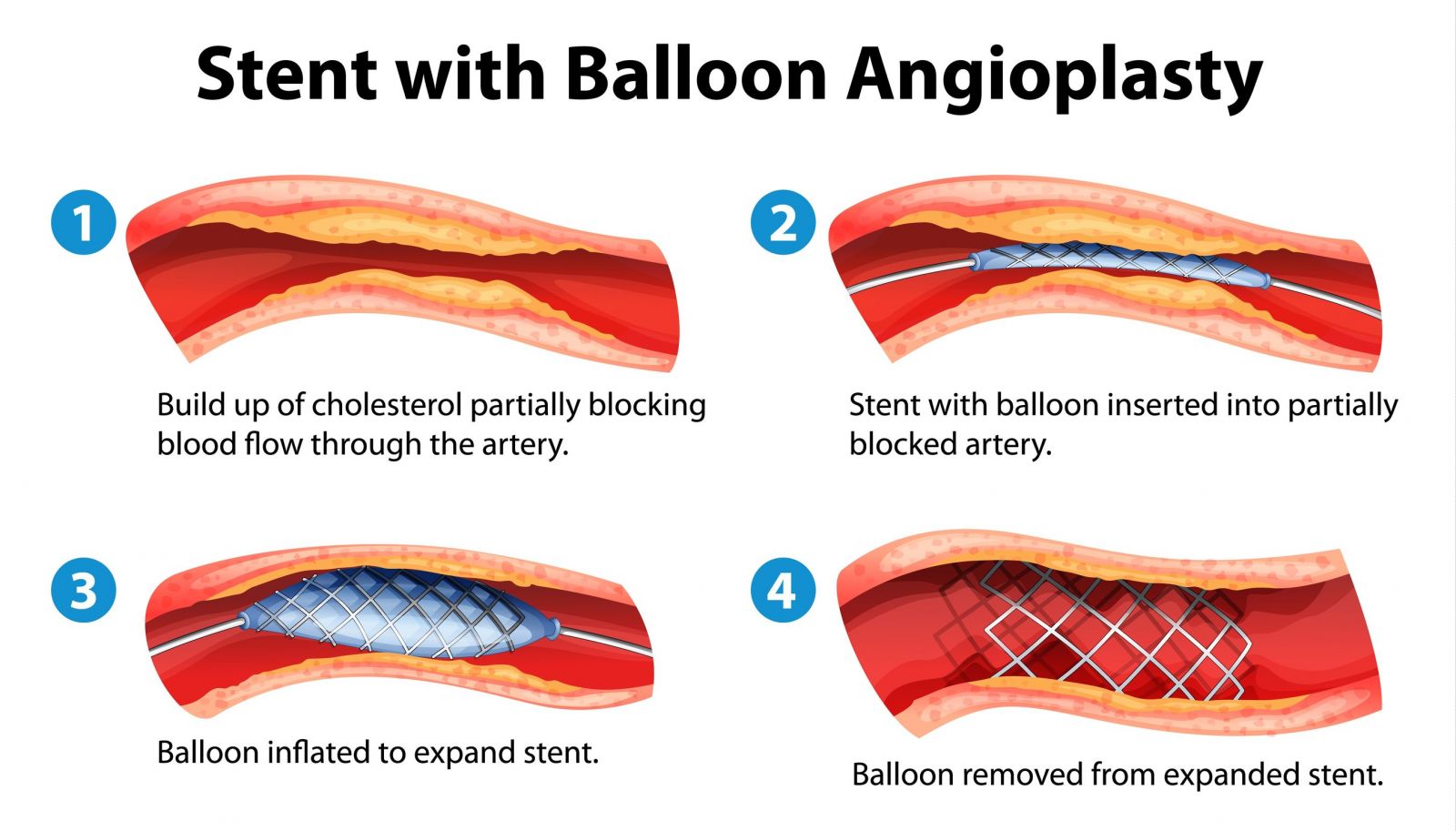
Fibrinolysis can open the blocked vessel in some instances and sometimes is an alternative to angioplasty. Agents such as tissue plasminogen activator (tPA) are used.
HOW WELL DO THE PATIENTS DO?
Outcome is variable and depends on the extent of myocardial damage. Often patients may die as a result of an MI or related complications.
WAS THERE A WAY TO PREVENT IT?
Preventing coronary artery disease (proper diet and exercise) will in part help prevent MI and other forms of heart disease.
WHAT ELSE ARE WE WORRIED ABOUT?
Complications with reperfusion may seem counterintuitive but can occur:
- Contraction band necrosis occurs when irreversibly damaged cells are reperfused. This leads to an influx in calcium, causing hyerpcontraction of myofibrils.
- Reperfusion injury can occur when the return of oxygen and inflammatory cells to the myocardium generates free radicals further damaging myocytes.
Arrhythmias: these can be fatal and are a common cause of death immediately during the MI (and even weeks afterward).
- Ventricular tachycardia can be a consequence of an old scar from an MI that causes abnormal conduction and a re-entry circuit into the ventricle (which ultimately causes rhythmic depolarization of the ventricles). Class IB sodium channel blockers can be used for this condition post MI.
Myocardial dysfunction is an obvious complication of MI (due to irreversible damage of the myocardium). Cariogenic shock can occur immediately with large MIs (low cardiac output and hypotension). Delayed heart failure is also possible (left sided heart failure is commonly caused by MI)
Mitral regurgitation can be a result of papillary muscle rupture after an MI. The chord tendineae no longer function to prevent regurgitation under these circumstances.
Ventricular free wall rupture can occur ~3-7 days after an MI. This will cause bleeding it not he pericardial, ultimately resulting in pericardial tamponade and death.
- A ventricular pseudoaneurysm can form when a free wall rupture is contained. This will decrease cardiac output, increase the risk of an arrhythmia, and mural thrombus emboli can form (~3-14 days post MI).
Fibrinous pericarditis: within the first week post MI, inflammation from the necrosis of the myocardium can increase vessel permeability and exudate formation. Directly caused by inflammation.
Ventricular aneurysm: a later complication (occurs months after MI) where wearing of the ventricular wall leads to dilation and fibrosis without rupture. This complication takes time simply because the fibrosis takes time to occur. The dilated fibrotic area of the heart cannot contract, and ultimately leads to congestive heart failure. Clots can also form along the wall of the ventricle (mural thrombus) due to the stasis of blood (these can become emboli).
Dressler syndrome (autoimmune fibrinous pericarditis): as the myocardium dies, cellular contents foreign tot he immune system are pleased and the body may develop an auto-immune response to them. This can lead to an autoimmune pericarditis ~1-2 months after the MI.
OTHER HY FACTS?
Coronary Vessel Anatomy:
- LAD is the most commonly occluded coronary vessel. Blockage of this vessel leads to infarction of the anterior wall/anterior septum of the left ventricle.
- Anteroseptal LAD: involves leads V1-V2
- Anteroapical (distal LAD): involves leads V3-V4
- Anterolateral (LAD): involves leadsV5-V6
- RCA is the second most commonly occlude vessel. Blockage of this vessel leads to infarction of the posterior wall/septum and papillary muscles of the left ventricle.
- Inferior (RCA): involves leads II, III, aVF
- LCX occlusion leads to infarction of the lateral wall of the left ventricle.
- Anterolateral (LCX): involves leads V5-V6
- Lateral (LCX): involves leads I, aVL
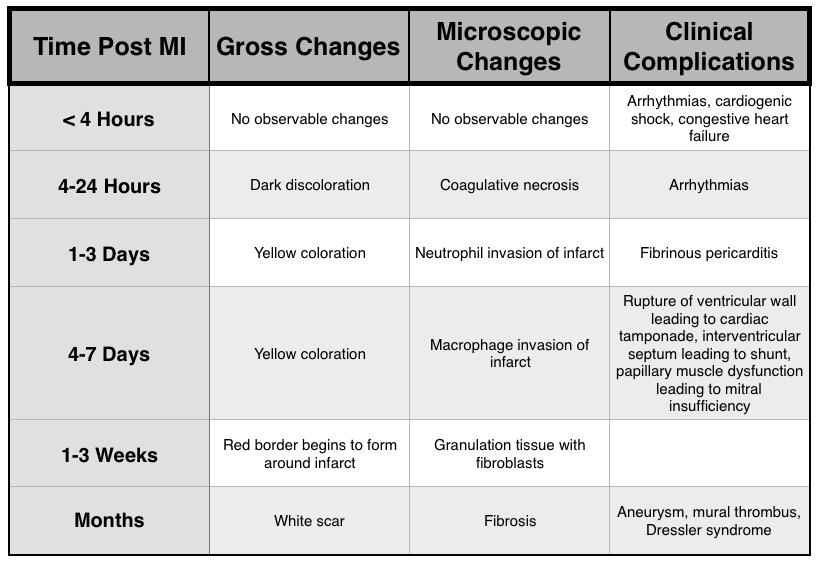
Gross/Microscopic Changes of MI:
Other:
- After an MI what was once functional myocardium is then replaced with scar tissue.
- Infarction generally involves the left and right ventricles (both atria are commonly spared).
- LDH, type 1 (lactate dehydrogenase) has been used in the past for diagnosing MI
FURTHER READING
Page Updated: 05.25.2016