Page Contents
- 1 WHAT IS IT?
- 2 WHY IS IT A PROBLEM?
- 3 WHAT MAKES US SUSPECT IT?
- 4 HOW DO WE CONFIRM A DIAGNOSIS?
- 5 HOW DO WE RULE OTHER DIAGNOSES OUT?
- 6 HOW DO WE TREAT IT?
- 7 HOW WELL DO THE PATIENTS DO?
- 8 WAS THERE A WAY TO PREVENT IT?
- 9 WHAT ELSE ARE WE WORRIED ABOUT?
- 10 OTHER HY FACTS?
- 11 ARCHIVE OF STANDARDIZED EXAM QUESTIONS
- 12 FURTHER READING
WHAT IS IT?
Hemochromatosis is a condition that is characterized by excess iron levels within the body. This condition can be hereditary (autosomal recessive condition often caused by mutations on the HFE gene) or can be acquired due to factors such as chronic transfusions (such as in patients with α-thalassemia or β-thalassemia).
WHY IS IT A PROBLEM?
Hereditary mutations will often lead to the dysfunction of hepcidin within the body. Given the role of hepcidin in stopping transport of iron from the cells of the intestine into the serum, this will result in constitutive absorption of iron from the gut.
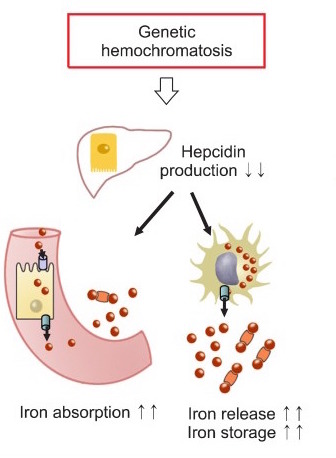
These high levels of iron will lead to iron deposition throughout tissues of the body (hemosiderosis). This iron will lead to free radical injury within the body which commonly leads to the classic triad of micronodular cirrhosis of the liver, diabetes mellitus, and skin pigmentation. The liver and pancreas are common sites of iron deposition, as is the heart (which can lead to cardiomyopathies).
WHAT MAKES US SUSPECT IT?
Risk factors: HLA A3, α-thalassemia, β-thalassemia
Most patients are asymptomatic however those with symptoms can complain of:
- Weakness
- Lethargy
- Impotence
- Arthritis (second and third MCP joints typical)
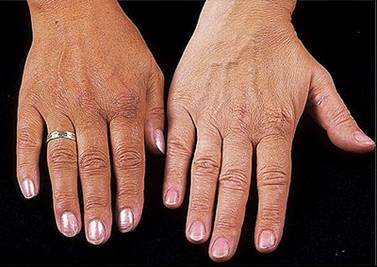
Skin hyperpigmentation (bronzing of skin) is often seen in this condition.
Testicular atrophy can be observed in patients (which can lead to gonadal dysfunction/impotence)
Abnormal liver function tests might be observed on routine screening.
HOW DO WE CONFIRM A DIAGNOSIS?
Serum lab results include the following:
- Increased ferritin
- Decreased TIBC
- Increased serum iron
- Increased % saturation of transferrin
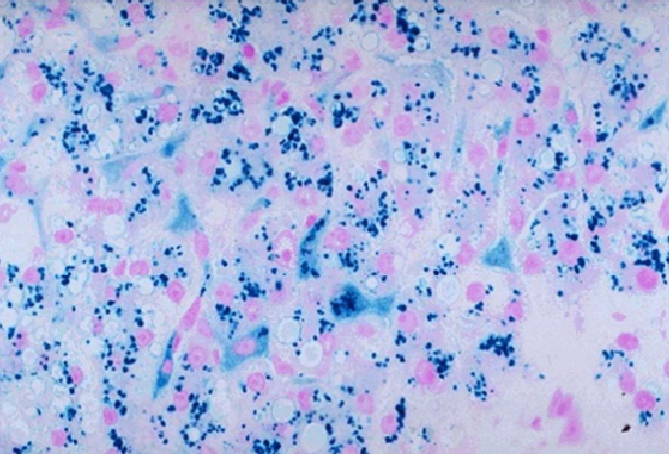
Liver biopsy will reveal the accumulation of brown pigment in hepatocytes. Using a Prussian blue stain iron can be distinguished in the biopsy (it stains blue) from the brown pigment of lipofuscin (normal byproduct of lipid metabolism found in liver).
Genetic screening can ultimately be diagnostic for patients with hereditary hemochromatosis.
HOW DO WE RULE OTHER DIAGNOSES OUT?
For hereditary hemochromatosis, genetic screening can be very useful in ruling out other diagnosis.
HOW DO WE TREAT IT?
Phlebotomy: patients are routinely undergo regular phlebotomies in order to maintain serum levels of iron within normal levels.
Iron chelation can also be performed using medications such as deferasirox, deferoxamine, deferiprone. These can be useful for patients who need chronic transfusions (i.e. thalassemia patients)
Menstruation: while not a “treatment” iron loss trough menstruation slows the progression of this condition in women.
HOW WELL DO THE PATIENTS DO?
Hereditary hemochromatosis patients can be well controlled with routine phlebotomy.
WAS THERE A WAY TO PREVENT IT?
While hereditary factors can not be prevented, patients who require chronic transfusions (α-thalassemia/β-thalassemia) should have their iron levels monitored closely, and can be prescribed iron chelators to avoid the acquisition of hemochromatosis.
WHAT ELSE ARE WE WORRIED ABOUT?
Dilated or restrictive cardiomyopathy can be caused by hemochromatosis and can lead to hear failure.
Hepatocellular carcinoma/hepatoma is associated with hemochromatosis.
OTHER HY FACTS?
“Bronze diabetes” is a common buzzword for this exam (skin pigmentation + diabetes in the classic presentation.
Common mutations include the C282Y (tyrosine for cysteine) or H63D on the HFE gene.
Vitamin C Excess can increase the risk of iron toxicity in patients with hemochromatosis.
Total body iron levels may reach 50g, which can be enough to set off metal detectors at airports.
Pseudogout is associated with hemochromatosis
HFE gene can stand for “high Fe”
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 03.23.2016