Page Contents
- 1 WHAT IS IT?
- 2 WHAT CAUSES IT?
- 3 WHY IS IT A PROBLEM?
- 4 WHAT MAKES US SUSPECT IT?
- 5 CLINICAL WORKUP
- 6 HOW DO WE RULE OTHER DIAGNOSES OUT?
- 7 HOW DO WE TREAT IT?
- 8 HOW WELL DO THE PATIENTS DO?
- 9 WAS THERE A WAY TO PREVENT IT?
- 10 WHAT ELSE ARE WE WORRIED ABOUT?
- 11 OTHER HY FACTS?
- 12 ARCHIVE OF STANDARDIZED EXAM QUESTIONS
- 13 FURTHER READING
WHAT IS IT?
Huntington disease (HD) is a genetically inherited (autosomal dominant) neurodegenerative condition that is characterized by the following neurotransmitter changes:
- Decreased GABA
- Decreased acetylcholine
- Increased dopamine
WHAT CAUSES IT?
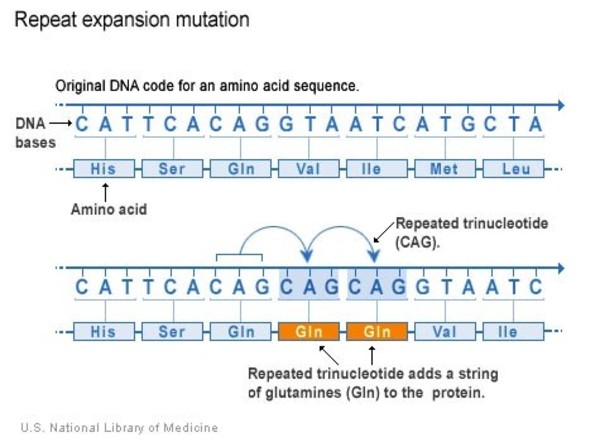
It is caused by the inheritance of trinucleotide repeats (CAG/glutamine) on the Huntington (HTT) gene (chromosome 4).
WHY IS IT A PROBLEM?
These trinucleotide repeat containing protein leads to neuronal death by binding NMDA-R receptors (as well as glutamate toxicity where neural cells are destroyed by glutamate over activation). The cells that digerati are the GABAergic neurons in the caudate nucleus of the basal ganglia. This combined with the neurotransmitter signaling changes seen above ultimately results in a movement disorder characterized by excessive voluntary movement. This is because the normal function of the basal ganglia is severely impaired.
Later in life (as more damage to the neurons occur) the patient may have their movements inhibited.
WHAT MAKES US SUSPECT IT?
Risk factors:
Family history
Initial presentation:
- Abnormal movements
- Mental status changes: dementia, aggression, depression
History Of Present Illness:
Onset: later in life, ages 20-50
Early signs: can include benign things such as dropping items
Personality changes: impulsive behavior and aggression may be observed by those around the patient.
Later stage disease can be characterized by stiffness, bradykinesia, difficulty to walk or eat.
Physical Exam:
General observations can demonstrate abnormal movements:
- Athetosis: these are slow writhing movements (that are “snake-like”) that are especially observed in the fingers.
- Chorea: sudden jerky movements down that a purpose (“dance-like”). These are non-repetitive, random movements the are not rhythmic.
HEENT:
- Abnormal eye movements: such as saccades (rapid movement between two points of focus) can be observed on physical exam.
CLINICAL WORKUP
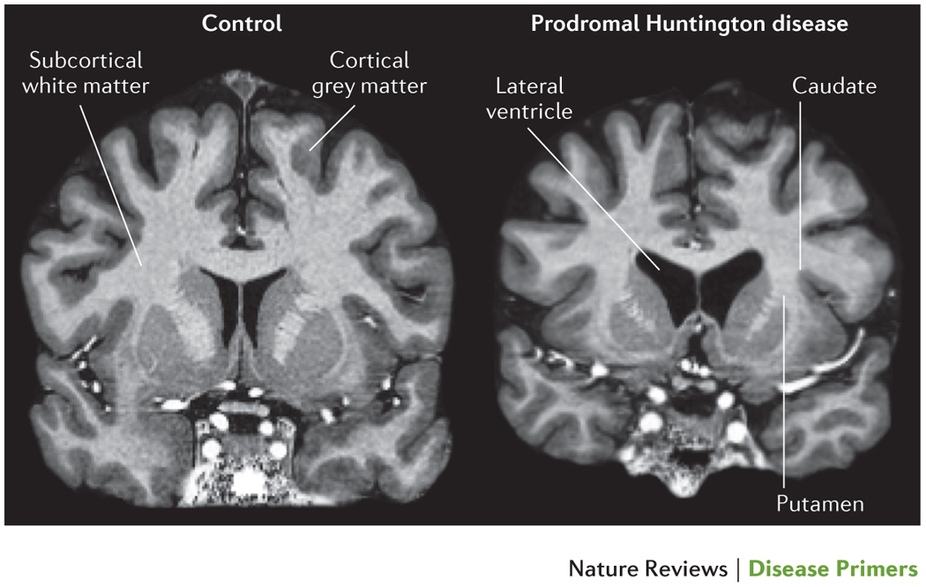
MRI can demonstrate atrophy of the striatum (caudate nuclei and/or putamen), also with ex vacua dilation of the frontal horns of the lateral ventricles.
Genetic testing: the diagnosis is confirmed by DNA analysis (≥ 36 CAG trinucleotide repeats in HTT gene)
HOW DO WE RULE OTHER DIAGNOSES OUT?
Genetic testing will be very useful in making a definitive diagnosis of the the condition.
HOW DO WE TREAT IT?
*No known curative treatment: patient symptoms (such as chorea) are treated as best as possible.
Tetrabenazine/reserpine: these medications inhibit the vesicular monoamine transporter (VMAT), which in turn limits dopamine vesicle packaging and release. Ultimately results in increased dopamine degradation.
Haloperidol: this is D2 receptor antagonist.
HOW WELL DO THE PATIENTS DO?
Median survival is about 15-20 years (source). There is no cure, all patients progress, their symptoms worsen, and this condition is ultimately fatal.
WAS THERE A WAY TO PREVENT IT?
While this genetic condition is not preventible, genetic counseling can serve to be very beneficial for patients who possess the condition and wish to avoid passing on the condition to their children.
WHAT ELSE ARE WE WORRIED ABOUT?
Suicide is a common cause of death given the seriousness of this condition and bleak outcome.
Depression is common in patients given the difficult nature of the condition.
OTHER HY FACTS?
Anticipation is a process by which these trinucleotide repeats expand generation to generation, and lead to the earlier onset of symptoms in future offspring.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 02.20.2016