Page Contents
WHAT IS IT?
Tuberous sclerosis is a genetic neurocutaneous disorder that also has multiple organ system involvement. It is characterized by numerous benign hamartomas (benign tumors) in the central nervous system, skin, and various organs.
WHAT CAUSES IT?
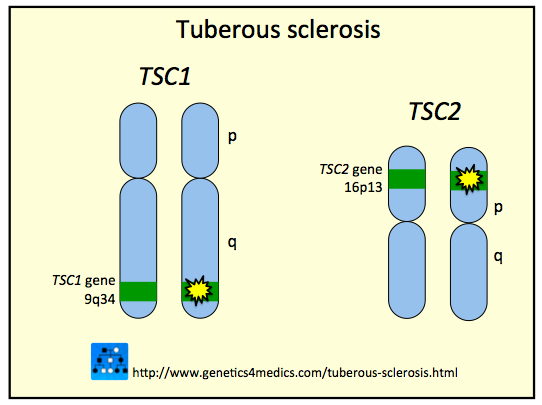
This condition is caused by an autosomal dominant mutation of the TSC1 (hamartin) or TSC2 (tuberin) gene. These genes are both tumor suppressor genes, so their decreased activity allows for tumor formation to occur much more readily.
WHY IS IT A PROBLEM?
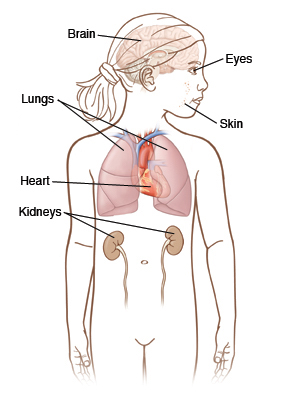
These widespread hamartomas can cause issues that are specific to the organ system involved (for example CNS involvement is likely responsible for the intellectual disability and seizures that are present in some patients).
WHAT MAKES US SUSPECT IT?
Risk factors
Family history of tuberous sclerosis
Initial Presentation
Incomplete penetrance: this genetic condition is not always completely penetrant (and has a variable presentation)
Common Chief Complaints:
- Facial growths/papules
- Intellectual disability
- Seizures
History Of Present Illness
Seizures are typically generalized convulsive seizures (infantile spasms in newborns-infants).
Intellectual disability is more likely the earlier the onset of this disease.
Physical Exam Findings
Dermatological exam: the below skin lesions can sometimes be enhanced by a Wood’s UV lamp.
- Angiofibromas (sebaceous adenomas) can be seen on the face/nose.
- Ash leaf spots are hypo pigmented macules on the skin (typically on trunk/extremities)
- Shagreen patches are associated with an increase in the incidence of subependymal astrocytomas and ungual fibromas.
Cardiac exam:
- Mitral regurgitation might be appreciated upon auscultation (in patients with cardiac tumors causing congestive heart failure).
CLINICAL WORKUP
Clinical diagnosis is common given characteristic physical exam findings explained above.
EKG may show findings that could support the presence of a rhabdomyoma of the heart (often in the apex of the left ventricle). This may be difficult to appreciate without a strong suspicion.
Chest X-ray may show pulmonary lesions and/or cardiomegaly that can be secondary to rhabdomyoma. This may be difficult to appreciate without a strong suspicion.
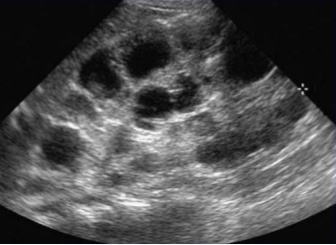
Renal ultrasound may show renal hamartomas/masses/or polycystic disease.
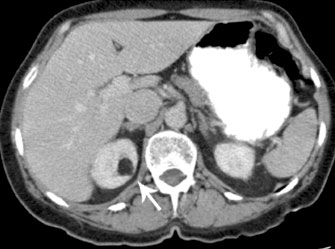
Renal CT scan can help show and diagnose angiomyolipomas.
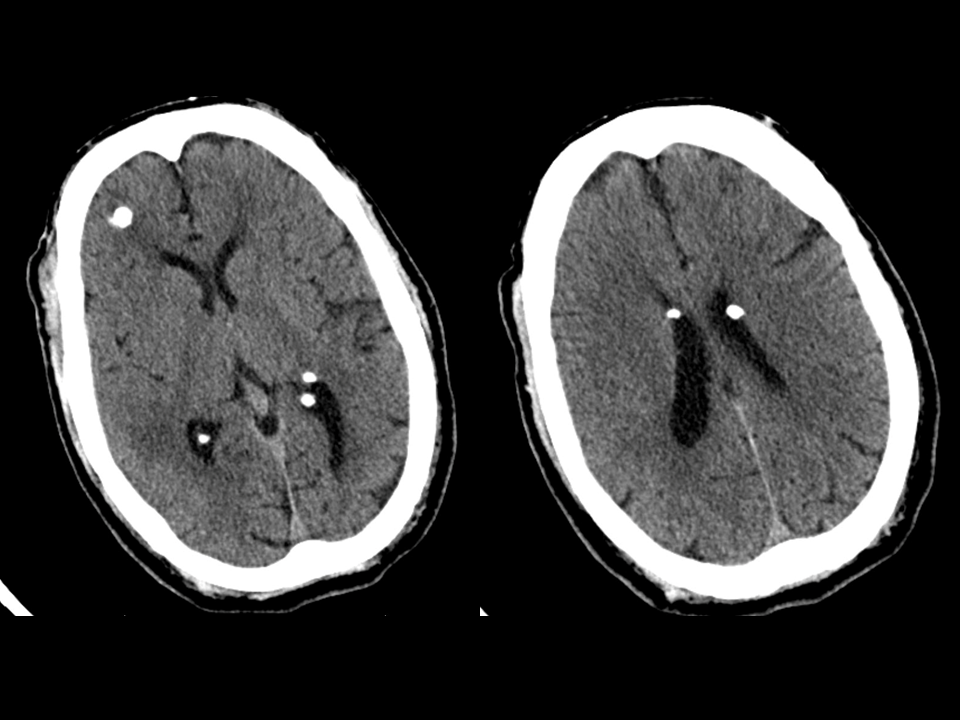
Head CT scan can reveal the pretense of calcified tubers within the cerebrum (particularly in the periventricular area).
Genetic testing can reveal the loss of function of the following tumor suppressor genes:
- TSC1: this codes for the Hamartin protein
- TSC2: this codes for the Tuberin protein
HOW DO WE TREAT IT?
Treatment will vary on the presentation/symptoms
Seizure control is often achieved with clonazepam or valproic acid. Surgical removal of tumors in the brain may be required.
- Infantile seizures are treated with ACTH
Other surgery may be required for other problematic tumors (in any of the affected locations).
HOW WELL DO THE PATIENTS DO?
This will vary on how penetrant the condition is in the affected patient (presentation and prognosis is very variable).
WAS THERE A WAY TO PREVENT IT?
This is a genetic condition with no clear method of prevention.
WHAT ELSE ARE WE WORRIED ABOUT?
Tumors in multiple different organs: these can all become malignant and can cause symptoms.
- Brain tumors such as giant cell astrocytomas. These can cause intellectual disability/seizures.
- Kidney tumors such as renal angiomyolipomas (very rarely renal cell carcinoma)
- Heart tumors such as cardiac rhabdomyomas. These can cause congestive heart failure.
- Retinal tumors which can include the mulberry tumors (arise from nerve head) and phakomas (which are round flat lesions located peripherally in the retina)
OTHER HY FACTS?
Infantile spasms (West syndrome) can occur within 6 months of birth due to this condition. This presents as generalized epilepsy.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 07.23.2016