Page Contents
WHAT IS IT?
β-Thalassemia is hematological condition that is caused by defective synthesis hemoglobin either due to decreased or absent synthesis of β-globin chains ( α-globin chain synthesis is normal).There are many mutations that can cause this condition (commonly point mutations in splice sites and promoters). Because there are only two copies of the β-globin genes, individuals either have β-thalassemia trait (minor, loss of one copy) or β- thalassemia major (Cooley anemia, loss of both copies).
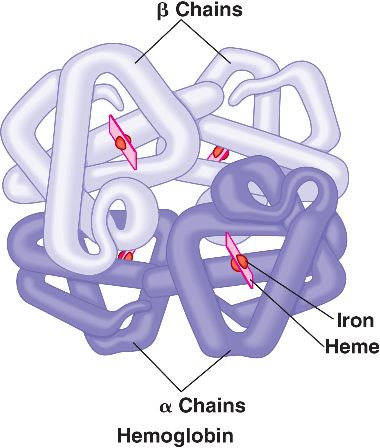
Semantics: complete loss of β-globin expression on an allele is referred to as βo and decreased expression of β-globin is deemed β+. Patients can be any combination of normal, low. or null expressers which explains the spectral phenotypes observed clinically.
WHY IS IT A PROBLEM?
In β-thalassemia excesses of free α-globin (relatively unstable) for tetramers that are destructive to the RBC’s leading to lack of their production (in the marrow). The body will try to compensate by increasing the physiological processes in place to promote RBC production, leading to a paradoxical exacerbation of the underlying pathology of this disease.
Ultimately the body will also try to compensate by extramedullary hematopoiesis (production of RBCs outside of the bone marrow, such as in the live or spleen) which will lead to clinical findings upon examination.
WHAT MAKES US SUSPECT IT?
Risk factors: mediterranean ancestry (i.e. Italian), African ancestry
Chipmunk facies: bone marrow expansion in facial bones will lead to a characteristic physical appearance of a “chipmunk face”
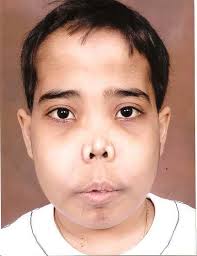
General symptoms of hemolytic anemia: fatigue, hepatospleanomegaly (due to extramedullary hematopoiesis), jaundice.

HOW DO WE CONFIRM A DIAGNOSIS?
Complete blood count: microcytic anemia will be seen on a CBC.
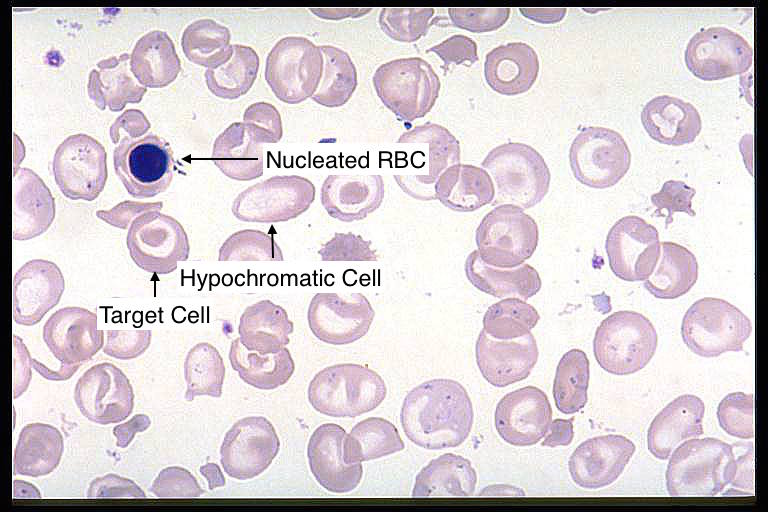
Blood smear: microcytic, hypochromic RBCs with target cells, as well as nucleated red blood cells can be seen on a peripheral blood smear.
Hemoglobin electrophoresis: elevated hemoglobin A2 (HbA2 > 3.5%) and also elevated fetal hemoglobin (HbF) can be seen on electrophoresis (in some milder cases only HbA2 is elevated, however in β-thalassemia major a significant increase in HbF is seen). Given the amount of free α-globin in RBCs, they will bind to any available delta-globin and gamma globin, producing HbA2 and HbF (respectively).
*Only patients with β-thalassemia major (genotype βo/βo) will have NO HbA because there is no expression of β-globin
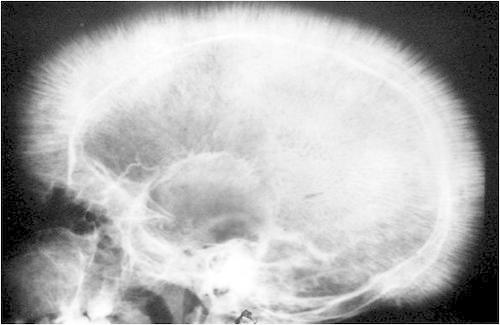
Cranial X-ray: while not completing diagnostic for only β-thalassemia, the skull will undergo bone marrow expansion leading to a characteristic “crew cut” appearance (looks as though there is hair on the x-ray that is standing on end).
HOW DO WE TREAT IT?
*Do not treat with iron! Patients will develop secondary hemochromatosis (iron overload).
Transfusions: patients are characterized by how often they (if at all) they need transfusions (transfusion dependent vs. independent patients). Transfusions are effective because they provide normal RBCs to the patient, however cary the risk of iron overload if done frequently.
Splenectomy: in patients with chronic splenomegaly, removal of the spleen may be required.
Bone marrow transplantation: only reserved for severe cases, this is the only curative method of treatment for the disease.
HOW WELL DO THE PATIENTS DO?
Prognosis is very variable depending on the type of β-thalassemia the patient has (as well as their dependency on transfusions).
WAS THERE A WAY TO PREVENT IT?
These genetic conditions are not preventable. Genetic counseling, genetic screening, and newborn screening are informative however and should be done to keep patients informed and educated regarding their carrier status, and risks associated with having children.
WHAT ELSE ARE WE WORRIED ABOUT?
Accumulation of iron due to chronic transfusions: giving transfusions often over long periods of time to thalassemias patients that require it is not benign and can lead to iron overload of patients. Giving iron chelators to patients at risk/experiencing iron overload can be beneficial because it helps the body exert excess iron.
Aplastic crisis: Increased ?risk of parvovirus B19–induced/infection induced aplastic crisis. An infection of RBC precursors will result in decreased RBC production.
OTHER HY FACTS?
HbS/β-thalassemia heterozygote: inheriting one copy of the sickle cell gene and one copy of another deficiency in β-globin (seen in β-thalassemi) can result in symptomatic patients (because no copies of normal β-globin are present). Patients present with mild/moderate sickle cell disease depending on the amount of β-globin production.
Fetal hemoglobin HbF is protective in this disease (which explains why fetuses are asymptomatic). Patients will not present with this condition until 6 months post birth (at which point the levels of fetal hemoglobin drop in newborns).
Page Updated: 01.08.2016