Page Contents
WHAT IS IT?
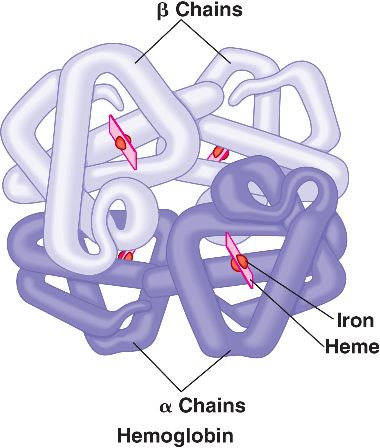
α-Thalassemia is hematological condition that is caused by defective synthesis hemoglobin either due to decreased or absent synthesis of α-globin chains ( β-globin chain synthesis is normal).There are multiple levels of genetic abnormalities that can cause α-Thalassemias (gene deletions are most common) because each cell carries 4 copies of the α-globin gene. If one copy is absent/deficient than the individual will be a silent carrier, two copies lead to the α- thalassemia trait, three defective genes lead to hemoglobin H (HbH) disease. Loss of all 4 copies is fatal during fetal development because alpha α-globin chains are required for the synthesis of fetal hemoglobin (HbF).
WHY IS IT A PROBLEM?
In α-thalassemia free β-globin chains (fairly stable), will aggregate and form abnormal hemoglobin H called HbH (β4). HbH is relatively insoluble, and forms intracellular precipitates that are referred to as heinz bodies. These heinz bodies will impair the deformability of the RBCs, leading to premature destruction of the cells by macrophages. Ultimately this can cause anemia due to moderate/severe hemolysis.
WHAT MAKES US SUSPECT IT?
Risk factors: African descent, Asian descent
*Patients with only 2 missing copies of the α-globin gene are generally asymptomatic
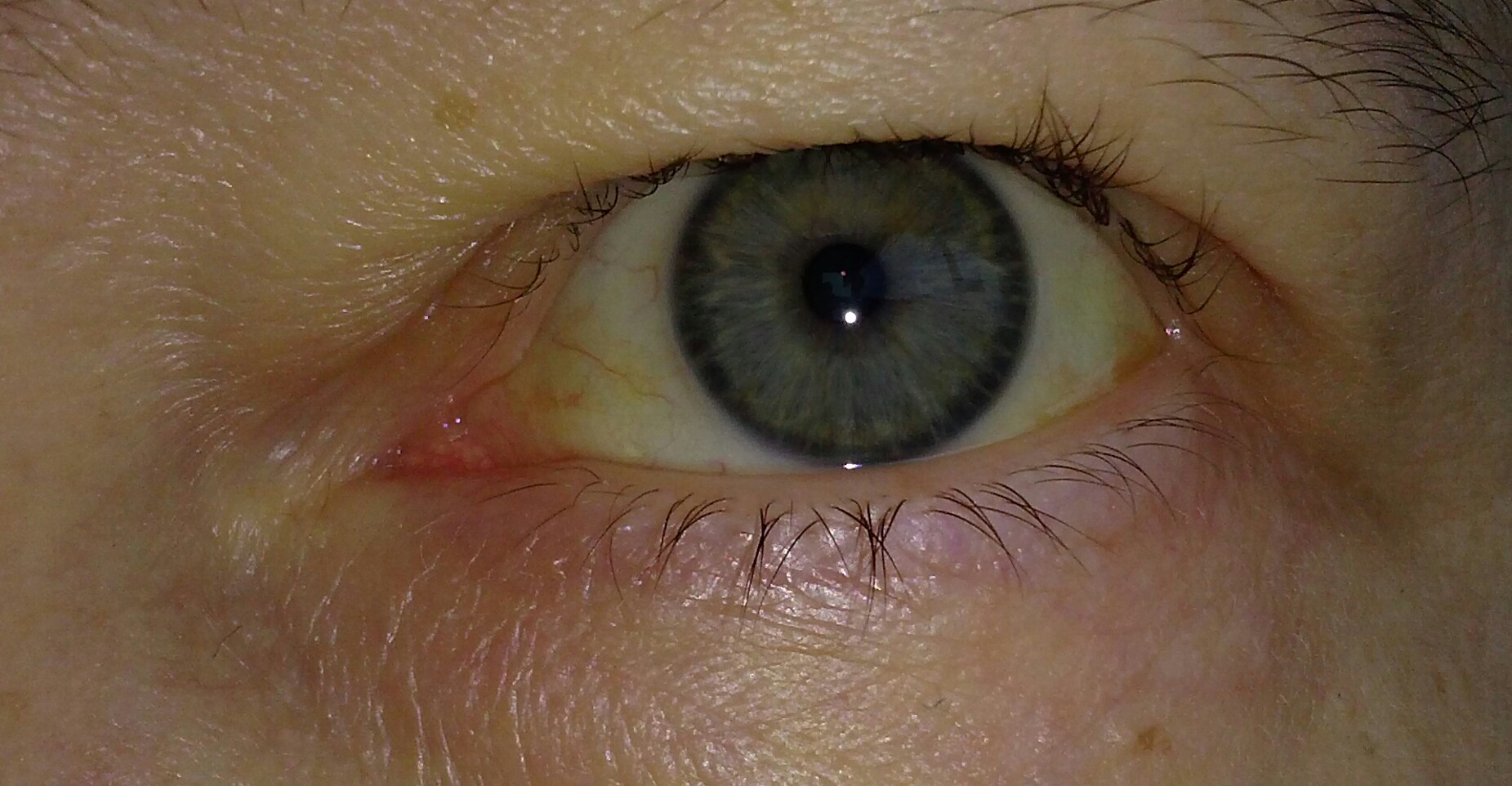
Patients with HbH disease can have a varying clinical presentation that can include mild jaundice, hepatosplenomegaly (felt on physical exam), as well as generalized symptoms of hemolytic anemia (fatigue, weakness, etc).
HOW DO WE CONFIRM A DIAGNOSIS?
Microcytic anemia: 1 missing copy of the α-globin gene will lead to slight microcytic anemia (decreased mean corpuscular volume/MCV). More missing copies will lead to more dramatic microcytic anemia.
Two-deletion α-thalassemias: normal or near-normal hemoglobin levels with microcytosis, occasional target cells, and an increased β/α synthesis ratio.
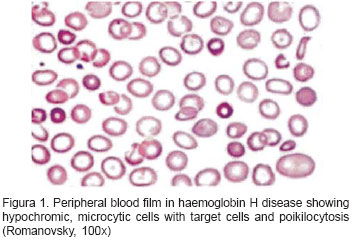
Hb H disease: microcytic, hypochromatic anemia seen on blood smear. Poikilocytes (abnormally shaped red blood cells) can also be seen.
HbH hemoglobin Analysis: variable amounts of HbH will be found in the patients blood. This will help in the diagnosis of this condition.
Molecular genetic testing can confirm diagnosis and determine deletion vs. nondeletion HbH disease (this could have prognostic value).
HOW DO WE TREAT IT?
**Avoid iron supplementation because iron-overlaod is common! These patients do not have a deficiency in iron! (exception is pregnant women with iron deficiency)
*Carriers and patients with only 1 dysfunction α-globin usually do not require treatment (exception of giving folate supplements to those who are deficient).
(HbH) disease treatment:
Supportive treatment: is usually common and includes folate acid supplements and treatment of infections.
Red Blood cell transfusions: patients with severe disease, viral infections causing aplastic crisis (ex. parvovirus B19), or pregnant women may require transfusions.
Iron chelation therapy: certain patients may require iron chelation therapy as well. These help remove iron from the body for patients who experience iron overload.
HOW WELL DO THE PATIENTS DO?
The prognosis is excellent for patients who are silent carries or only have 1 dysfunction α-globin gene (due to being asymptomatic)
The prognosis for patients with HgH disease is variable given diverse phenotypes and disease severity.
WAS THERE A WAY TO PREVENT IT?
These genetic conditions are not preventable. Genetic counseling, genetic screening, and newborn screening are informative however and should be done to keep patients informed and educated regarding their carrier status, and risks associated with having children.
WHAT ELSE ARE WE WORRIED ABOUT?
Hydrops fetalis: Inheritance of 4 dysfunctional α-globin genes is fatal and will lead to hydrops fetalis (death of the fetus in utero). Counseling patients who carry dysfunctional α-globin about this condition might be worthwhile in patients who are trying have children.
Iron overload: While not very common in a setting without iron supplementation, this condition (especially for patients who undergo chronic RBC transfusion) can lead to iron overload. One should aways look out for typical symptoms of iron overload with include:
- tiredness/weakness
- loss of libido
- weight loss
- abdominal pain
- joint aches or pain
Serum iron studies (serum transferrin and ferritin) can be used to assess the circulation levels of iron within a patients blood (and can confirm iron overload).
OTHER HY FACTS?
Inheritane of a single α-thalassemia gene might give protection against falciparum malaria.
Page Updated: 01.06.2016