Page Contents
WHAT IS IT?
Thrombotic thrombocytopenic purpura (TTP) is a condition of excessive clotting caused by a deficiency in the ADAMTS13 enzyme which cleaves von Willebrand factor (vWf), a large protein involved in blood clotting. This deficiency is rarely congenital/genetic, and usually is acquired as an autoinhibitory IgG antibody. It is important to realize that this deficiency combined with a second trigger (specifics drugs, anti platelet agents, lupus, infection, etc) is ultimately the cause for this condition.
WHY IS IT A PROBLEM?
Give that the ADAMTS13 normally is involved in antagonizing clot formation, its inhibition sensitizes patients to clot development. Especially in combination with a second trigger (such as anti platelet therapy) micro thrombi will form and then occlude small vasculature within the body (leading to a host of problems/the physical presentation of this condition).
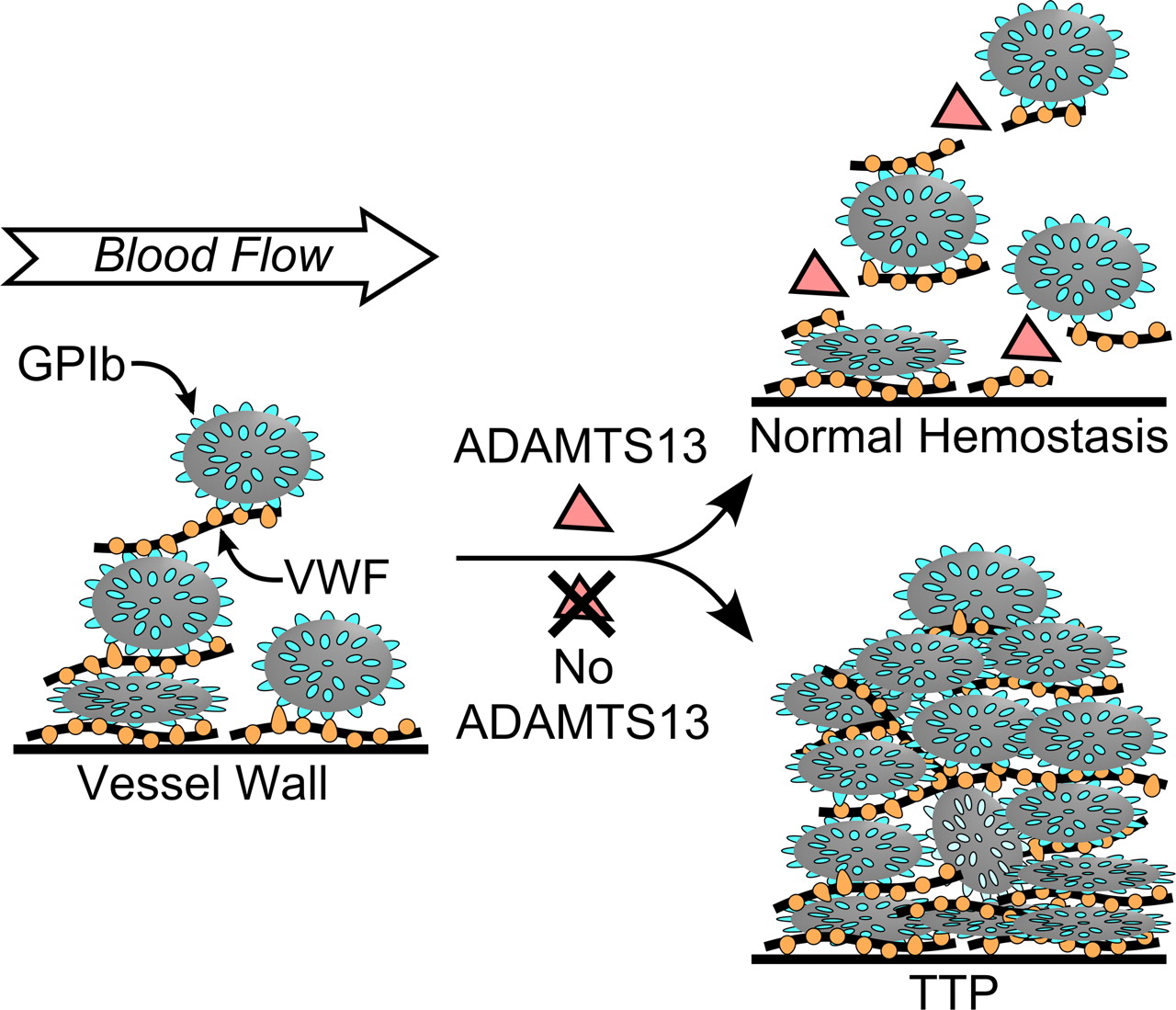
Ultimately the formation of these clots will cause a pentad of symptoms which include: neurological issues, renal symptoms, fever, thrombocytopenia (due to usage of platelets), and microangiopathic hemolytic anemia (erythrocytes are physically destroyed by being pushed through the fibrin networks of clots).
WHAT MAKES US SUSPECT IT?
Risk factors: female
Chief Concerns: weakness, nausea, diarrhea, anorexia, vomiting, fever, weakness/fatigue (common symptom of anemia)
Transient Neurological Defects: generalized headache, seizures, confusion, coma, altered sensory motor defects.
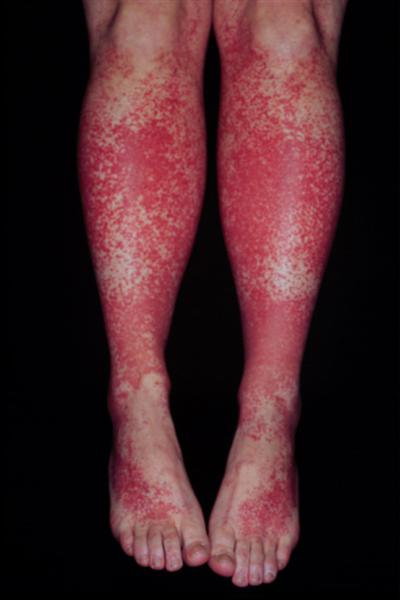
Skin findings: certain skin findings (with the above clinical presentation) can help suggest TTP
Purpura: this is a rash of purple/red color that appears on the skin. It is caused by internal bleeding from small vessels in the body and will not blanch (turn white when pressure is applied, indicating that blood is extravascular and can not be “pushed” away).
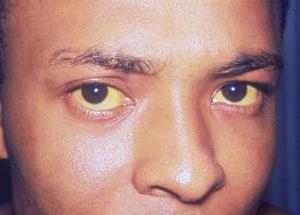
Jaundice: increased unconjugated bilirubin in the blood (due to hemolysis) will result in physical findings of jaundice.
HOW DO WE CONFIRM A DIAGNOSIS?
**This condition should not have coagulopathy (increased PT and PTT). If you see these consider DIC strongly!
Decreased haptoglobin: haptoglobin is a hormone that binds free hemoglobin in the blood. Once bound, it (and the conjugated hemoglobin) is removed by the spleen from circulation. Because TTP causes intravascular hemolysis, haptoglobin levels will be decreased in patients.
Increased Lactate Dehydrogenase (LDH): this is an enzyme found in great quantities in blood cells (also found in a wide variety of cells) that is elevated in hemolytic anemias.
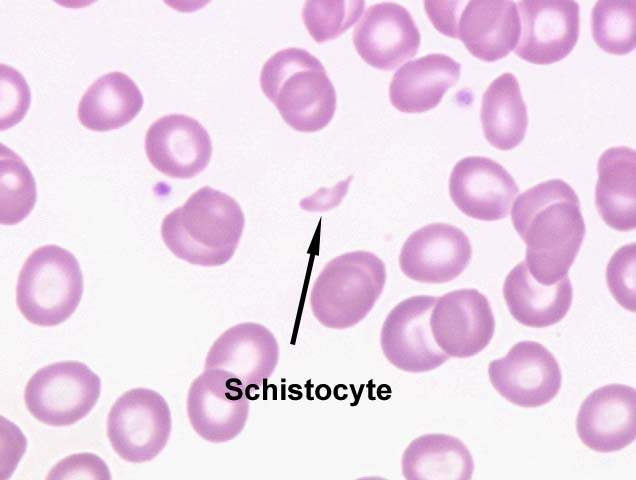
Schistocytes on smear: these are fragmented erythrocytes caused by the microangiopathic hemolytic anemia.
Decreased ADAMTS13 activity: while not done routinely for the initial diagnosis, to ultimately confirm the diagnosis of TPP ADAMTS13 enzyme activity can be directly measured.
HOW DO WE TREAT IT?
Plasmapheresis: is often the firstling therapy for patients with TPP. This is essentially a blood plasma transfer (where the causal antibodies are removed from the plasma of the patient and replaced with “normal” plasma).
Immunosuppressants: if plasmapheresis is not effective for certain patients, immunosuppressants such as steroids can be used.
HOW WELL DO THE PATIENTS DO?
Plasmapheresis is effective in ~80-90% of patients (source) and ultimately patients do quite well with this condition if managed properly.
WAS THERE A WAY TO PREVENT IT?
While some of the secondary triggers to a TPP event are avoidable (i.e. prescription of anti-platelet drugs in a patient with a past history of TPP), there are not clear preventions for acquiring the deficiency in ADAMTS13.
WHAT ELSE ARE WE WORRIED ABOUT?
Renal failure: patients with TPP (especially those induced by the drug quinine) can be at risk for renal failure (given deposition of thrombi in the vasculature of the kidney. Assessing creatinine levels can help monitor kidney function.
OTHER HY FACTS?
Page Updated: 01.06.2016