Page Contents
WHAT IS IT?
Friedreich ataxia is a neurodegenerative disorder that involves the spinal cord. Most notably the spinocerebellar tracts are involved however the dorsal columns, dorsal root ganglia, and lateral corticospinal tracts can also be affected.
WHAT CASUES IT?
This is an autosomal recessive disorder in which trinucleotide repeats (GAA) leads to decreased transcription of the frataxin protein (FXN gene).
WHY IS IT A PROBLEM?
Frataxin is essential for mitochondrial function and is concentrated in neurological tissues. Loss of its function causes mitochondrial dysfunction because of free radical damage from cytoplasmic iron buildup which ultimately causes damage to neurological tissues. Most specifically the spinocerebellar tracts undergo degeneration although other structures in the spinal cord can also be affected. Cerebellar dysfunction (resulting from the loss of spinocerebellar tract function) is responsible for the symptoms of ataxia that patients experience.
WHAT MAKES US SUSPECT IT?
Risk factors:
Caucasian, family history of cerebellar ataxia
Chief Complaints:
- Gait instability
- Clumsiness
- Frequent falling
Physical Exam:
General observations:
- Dysarthria can be noticed in some patients
- Wide based gait is a common finding
HEENT exam:
- Nystagmus
Neurological findings (variable): degeneration of multiple spinal cord tracts leads to lost vibratory sense, proprioception, muscle weakness in the lower extremities, and loss of deep tendon reflexes.
Musculoskeletal exam:
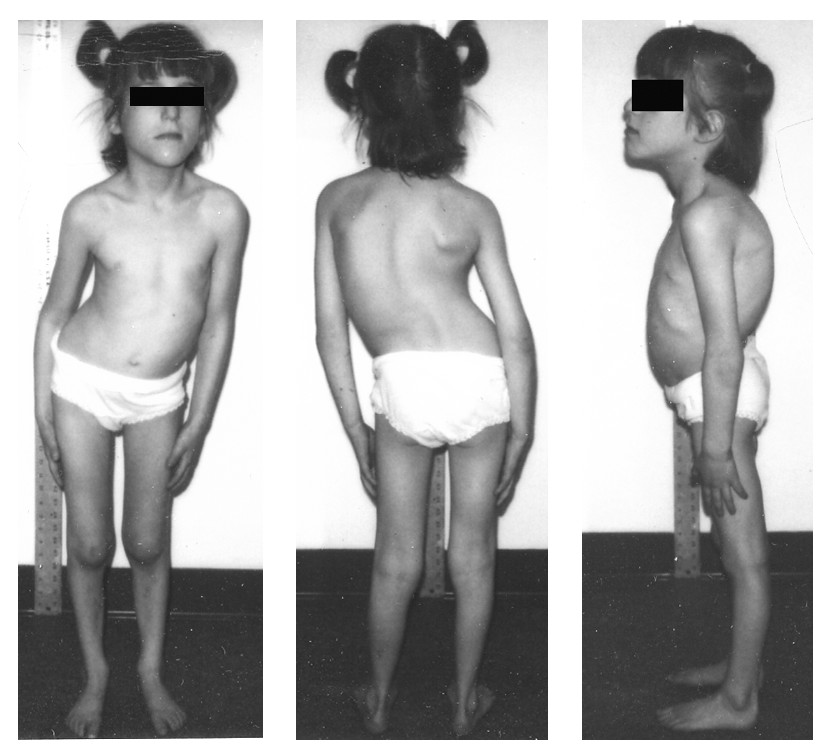
- Kyphoscoliosis is a characteristic feature of this condition.
- High plantar arches (pes caves) can be observed
- Hammer toe deformity can also be present.
CLINICAL WORKUP
MRI of the brain can show cerebellar atrophy.
MRI of the spine can show atrophy of the spinal cord.
HOW DO WE CONFIRM A DIAGNOSIS?
Given the above clinical suspicion, definitive diagnosis is done by genetic testing and confirming and abnormality in the FXN gene (increased trip-nucleotide repeats).
- normal individuals have 5-33 triplet
- patients with Friedreich ataxia have about 66-1,700 triplets (usually 600-1,200), source
HOW DO WE TREAT IT?
No disease modifying treatments are available as of now, and support for drug therapy is limited.
Repeat EKGs every 1-2 years are recommended to monitor patients for likely heart conditions.
- T-wave inversions can be observed as heart conditions develop.
HOW WELL DO THE PATIENTS DO?
Usually slowly progressive, but progression may be variable (source)
- mean time from onset to requiring a wheelchair for mobility about 10 years
- most patients wheelchair-dependent by their late teens or early twenties
- mean age of death 37 years
*Death is usually secondary to hypertrophic cardiomyopathy.
WAS THERE A WAY TO PREVENT IT?
This is a genetic condition without a clear prevention.
Genetic screening is possible for those who are at risk earlier on (family history of cerebellar ataxia) however can be controversial.
WHAT ELSE ARE WE WORRIED ABOUT?
Patients often develop heart disease (such as conduction defects or cardiomyopathy) and diabetes.
Hypertrophic cardiomyopathy is associated with this condition.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
Page updated: 08.15.2016