Page Contents
- 1 WHAT IS IT?
- 2 WHY IS IT A PROBLEM?
- 3 WHAT MAKES US SUSPECT IT?
- 4 HOW DO WE CONFIRM A DIAGNOSIS?
- 5 HOW DO WE RULE OTHER DIAGNOSES OUT?
- 6 HOW DO WE TREAT IT?
- 7 HOW WELL DO THE PATIENTS DO?
- 8 WAS THERE A WAY TO PREVENT IT?
- 9 WHAT ELSE ARE WE WORRIED ABOUT?
- 10 OTHER HY FACTS?
- 11 ARCHIVE OF STANDARDIZED EXAM QUESTIONS
- 12 FURTHER READING
WHAT IS IT?
DiGeorge Syndrome: is a genetic condition where a portion of the 22nd chromosome (22q11) is deleted. Most deletions arise de novo with same frequency in all populations, however it is possible that there is some genetic predisposition.
WHY IS IT A PROBLEM?
Losing this portion of the chromosome will cause various deficits/defects in the patient including: cardiac defects, immunodeficiency, hypoparathyroidism, palatal defects, and learning disabilities.
WHAT MAKES US SUSPECT IT?
Risk factors: family history
Chief concern (CC) most common presentations include history of some combination of: Congenital heart disease, immune deficiency, palatal abnormalities, nasal regurgitation, and/or hypernasal speech, developmental delays and/or learning difficulties. Behavior and/or psychiatric issues common.
Palate/facial abnormalities are common in patients with DiGeorges.
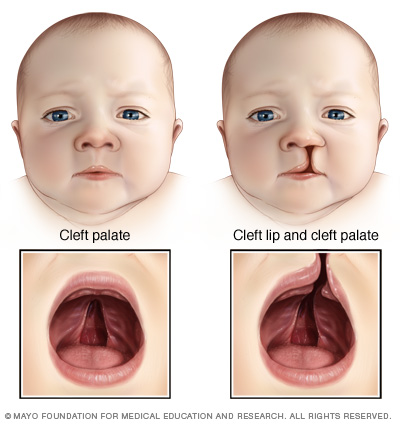
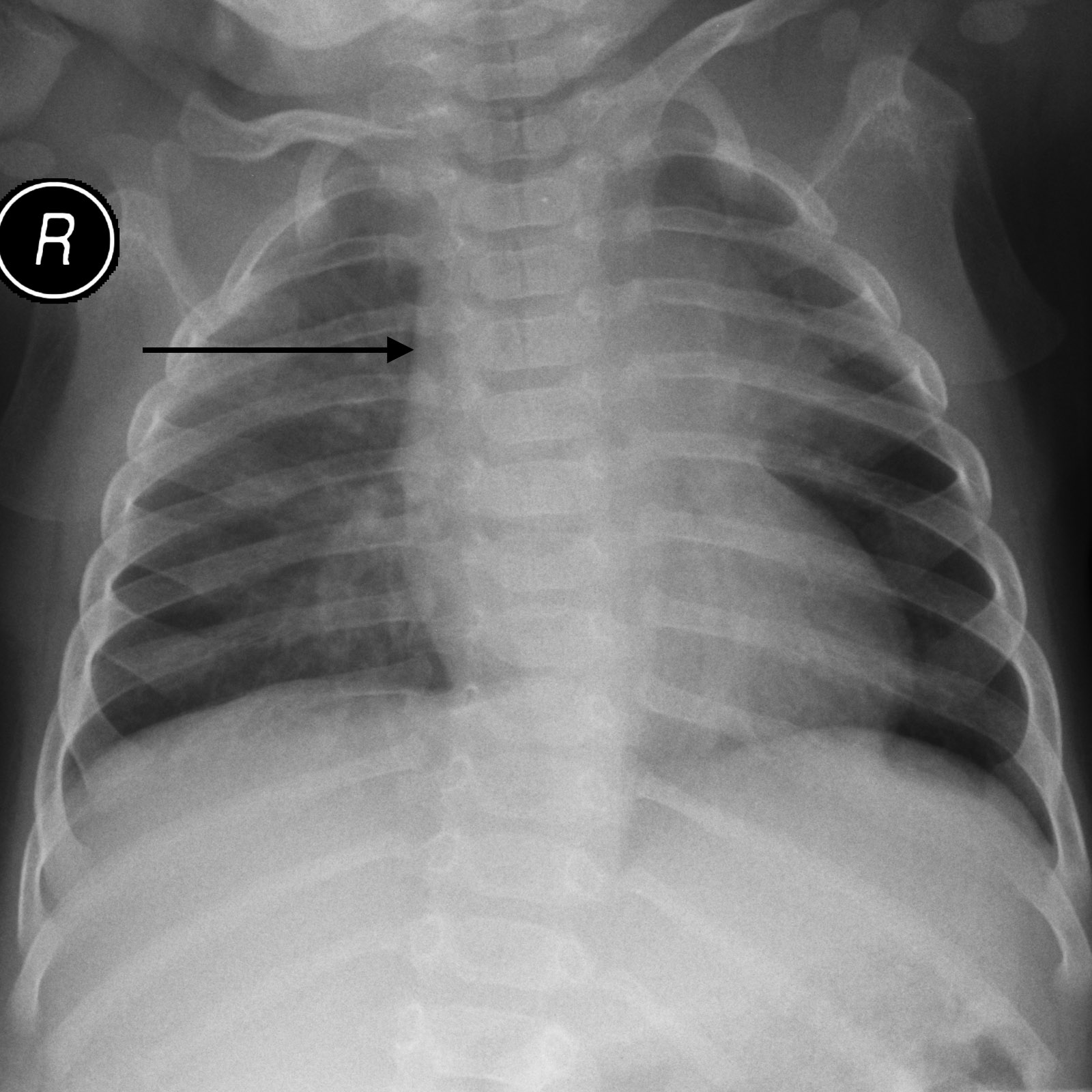
HOW DO WE CONFIRM A DIAGNOSIS?
Absent thyme shadow can be observed on X-ray. While not diagnosis on its own, this finding will help build the diagnosis.
Genetic testing: diagnosis confirmed by detection of deletion in chromosome 22q11 (genetic testing/FISH)
HOW DO WE RULE OTHER DIAGNOSES OUT?
Genetic sequencing ultimately can rule out the possibility of other diagnoses
HOW DO WE TREAT IT?
Treatments effective for individual clinical findings include:
- calcium supplementation with vitamin D for patients with hypocalcemia
- cardiac surgery for patients with congenital cardiac defects
- thymus transplant to reconstitute T-cell function
- surgery to repair palate defects and/or velopharyngeal insufficiency
HOW WELL DO THE PATIENTS DO?
Patients with 22q11.2 may have decreased life span with increased risk of sudden death (often due to congenital hear defects)
> 90% of all deaths attributed to 22q11.2 syndrome associated with congenital heart defect
WAS THERE A WAY TO PREVENT IT?
No this is a congenital condition (however prenatal ultrasound screening is possible)
WHAT ELSE ARE WE WORRIED ABOUT?
Infection: absence of thymus/normal T-cell function predisposes patients to infection.
Hypocalcemia: Absence of parathyroids causes conditions associated with hypocalcemia/low PTH.
OTHER HY FACTS?
**CATCH-22 (Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, and Hypocalcemia) and 22nd chromo deletion.
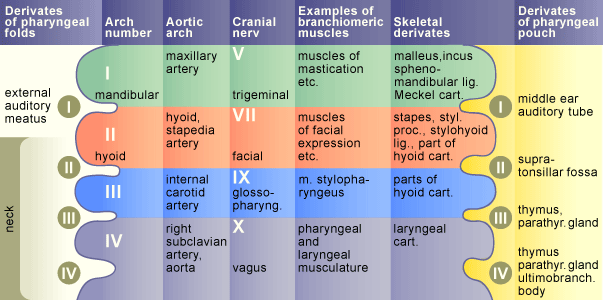
In DiGeorges syndrome the 3rd and 4th pharyngeal pouches do not develop properly (leading to loss of thymus and parathyroids)
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 02.02.2016