Page Contents
- 1 WHAT IS IT?
- 2 WHAT CAUSES IT?
- 3 WHY IS IT A PROBLEM?
- 4 WHAT MAKES US SUSPECT IT?
- 5 HOW DO WE CONFIRM A DIAGNOSIS?
- 6 HOW DO WE RULE OTHER DIAGNOSES OUT?
- 7 HOW DO WE TREAT IT?
- 8 HOW WELL DO THE PATIENTS DO?
- 9 WAS THERE A WAY TO PREVENT IT?
- 10 WHAT ELSE ARE WE WORRIED ABOUT?
- 11 ARCHIVE OF STANDARDIZED EXAM QUESTIONS
- 12 FURTHER READING
WHAT IS IT?
Amyotrophic lateral sclerosis (ALS) is commonly known as Lou Gehrig disease. It is a degenerative disorder of the upper and lower motor neurons of the corticospinal tract.

WHAT CAUSES IT?
Some familial cases of ALS have been linked to a defect in the superoxide dismutase 1 gene (SOD1). This gene codes for an antioxidant that normally neutralizes superoxide (a toxic free radical). When SOD1 is dysfunctional, it seems free radial injury will damage neurons.
WHY IS IT A PROBLEM?
In this condition both the anterior motor horn and the lateral corticospinal tract will degenrate leading to severe loss of muscle function (and both upper/lower motor neuron signs). It will eventually lead to respiratory compromise and then death.
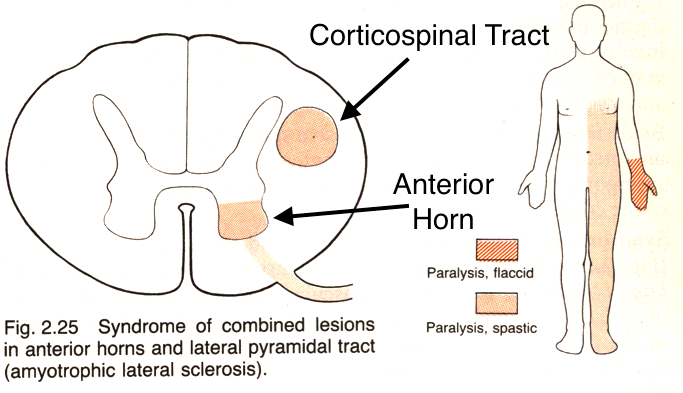
WHAT MAKES US SUSPECT IT?
Risk factors: family history, male sex, onset 40-80s.
Weakness/Atrophy in hands is one of the earliest signs of ALS.
- Weakness can be preset elsewhere and is generally asymmetric.
Other symptoms can include:
- Dysphagia
- Dysarthria
- Tongue atrophy/fascinations
- Respiratory compromsie
**Both upper motor neuron and lower motor neuron signs can be seen in ALS**
Upper motor neuron signs:
- Weakness/paralysis
- Increased reflexes
- Positive Babinski sign
- Increased muscle tone (spasticity)
Lower motor neuron signs
- Weakness/paralysis
- Fasciculations
- Muscle atrophy
- Decreased muscle tone
- Impaired reflexes
Absence of the following is most consistent with ALS:
- Sensory deficits
- Oculomotor deficients
- Fecal incontinence
- Changes in cognition
HOW DO WE CONFIRM A DIAGNOSIS?
Clinical diagnosis is often made based upon the above presentation. Presence of both upper and lower motor neuron signs in at least 1 limb can help very strongly suggest the diagnosis of ALS.
Nerve conduction studies/EMG will reveal widespread enervation and fibrillation potentials.
CT/MRI of the spine will often be done in order to rule out the presence of structural lesions that could explain the patient’s symptoms.
HOW DO WE RULE OTHER DIAGNOSES OUT?
The combination of both upper motor neuron and lower motor neuron signs should make one consider the possibility of ALS.
The lack of sensory impairment distinguishes ALS from syringomyelia.
HOW DO WE TREAT IT?
Riluzole (glutamate antagonist) treatment has been shown to modestly improve patient survival. It works to decrease the level of presynaptic glutamate release.
HOW WELL DO THE PATIENTS DO?
This is a fatal condition with a very poor prognosis (mean survival is 5 years from the initial diagnosis)
WAS THERE A WAY TO PREVENT IT?
N/A
WHAT ELSE ARE WE WORRIED ABOUT?
Loss of respiratory function will occur when muscle weakness has progressed enough and patients will die as a result.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated: 07.12.2016