Page Contents
WHAT IS IT?
Cystic fibrosis (CF) is an autosomal recessive disorder that is caused by a deficiency/mutation in the Cystic fibrosis transmembrane conductance regulator (CFTR) gene. It is characterized by the loss of chloride moment that manifests in multisystem disease.
WHY IS IT A PROBLEM?
Issues with the CFTR gene cause a decrease in chloride transport across membranes => decreased water flow following chloride ions => increased mucus persistence in areas like the pulmonary tracts=> associated symptoms
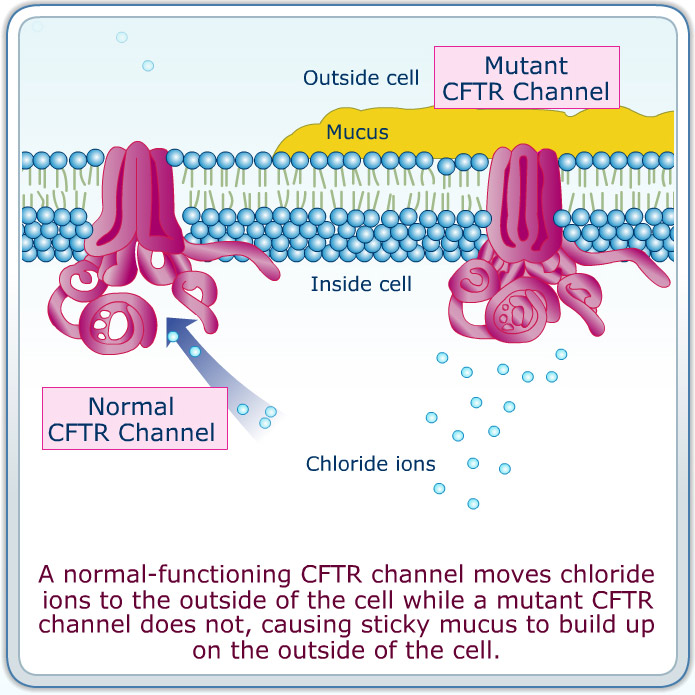
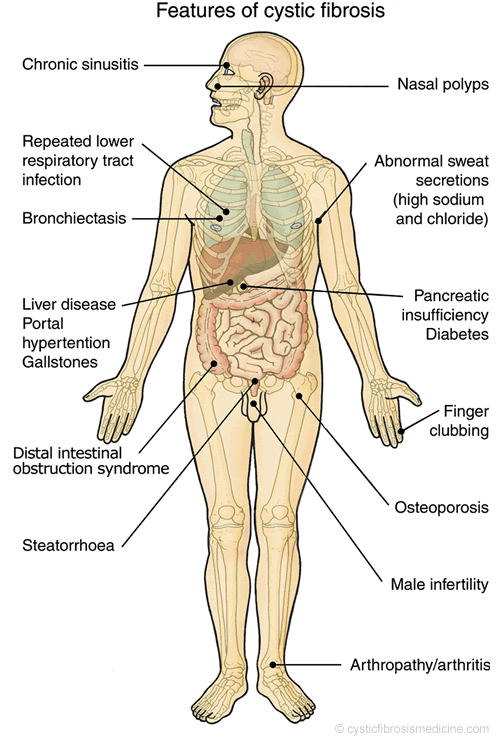
It affects the upper and lower airway, lungs, exocrine pancreas, small and large intestines, hepatobiliary system, salivary and sweat glands, and vas deferens.
WHAT MAKES US SUSPECT IT?
Risk factors: family history, caucasian
Chief complaint: cough with sputum, recurrent respiratory symptoms, salty skin
Pulmonary exam: crackles and/or wheezing can be heard on the exam.
HOW DO WE CONFIRM A DIAGNOSIS?
CF diagnosed by ≥ 1 characteristic clinical feature of CF, history of CF in sibling, or positive newborn screen PLUS laboratory evidence of abnormality in CF transmembrane conductance regulator (CFTR) gene or protein.
Acceptable lab evidence (1 of following)
• 2 disease-causing mutations in CFTR gene by molecular genetic testing
• 2 quantitative pilocarpine iontophoresis sweat chloride values > 60 mEq/L (60 mmol/L)
• Transepithelial nasal potential difference measurements characteristic of CF
*It is important to keep in mind that while sweat chloride levels are a common way to diagnose CF, a considerable number of patients might not have abnormal values.
**Another important point regarding genetic testing is that there are MANY genetic causes of the disease and certain screening exams only look at the most common mutations. If a patient is negative on an initial screening but the suspicion of CF is still high either a more in depth screening or full sequencing of the CFTR may be indicated.
HOW DO WE TREAT IT?
N-Acetylcysteine: this is a medication that loosens mucus plugs by cleaving disulfide bonds within mucus glycoproteins.
Dornase alfa (Pulmozyme): recombinant human DNase (rhDNase) therapy associated with improvement in lung function (thought to clear leukocytic debris).
Inhaled hypertonic saline: this treatment lacks direct evidence however it might provide benefit to patients.
Ivacaftor: used for patients with at least one G551D mutation. This mutation causes inproper opening of the CFTR channel protein, and Ivacaftor binds the CFTR and acts as a channel “potentiator” (essentially it allows the channel to open and operate properly).
Antibiotics/Antifungals: while the underlying condition of CF can not be treated with this therapy, the frequent pulmonary infections that are common should be treated with pathogen specific antibiotics. Common causes of pulmonary infection are: Pseudomonas aeruginosa, S. aureus, S pneumoniae, and Aspergillus fumigatus.
HOW WELL DO THE PATIENTS DO?
Predicted median survival 41.1 years in United States (source)
WAS THERE A WAY TO PREVENT IT?
While the genetic cause of CF is not preventable, newborn screening for CF is implemented in all of the U.S. (however some less common mutations will not be picked up on screens).
WHAT ELSE ARE WE WORRIED ABOUT?
Pancreatic insufficiency: certain individuals with CF are at risk for developing pancreatic insufficiency, and these patients should be given supplemental fat soluble vitamins (because the lack of proper pancreatic function will cause deficiency in fat soluble vitamins).
Bronchiectasis: CF is the most common cause of this respiratory loss of elastic tissues.
Meconium ileus: In cystic fibrosis, meconium plug obstructs intestine, preventing stool passage at birth.
Male infertility: male patients with CF are almost always infertile because of improper development of the vas deferens.
OTHER HY FACTS?
ΔF508 mutation is the most common cause of CF: a 3-nucleotide deletion on chromosome 7 coding for a phenylalanine residue The mutation affects the CFTR channels by causing them to be prematurely degraded in the cell’s Golgi apparatus.
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
Page Updated: 11.06.2016