Page Contents
- 1 WHAT IS IT?
- 2 WHY IS IT A PROBLEM?
- 3 WHAT MAKES US SUSPECT IT?
- 4 HOW DO WE CONFIRM A DIAGNOSIS?
- 5 HOW DO WE RULE OTHER DIAGNOSIS OUT?
- 6 HOW DO WE TREAT IT?
- 7 HOW WELL DO THE PATIENTS DO?
- 8 WAS THERE A WAY TO PREVENT IT?
- 9 WHAT ELSE ARE WE WORRIED ABOUT?
- 10 OTHER HY FACTS?
- 11 ARCHIVE OF STANDARDIZED EXAM QUESTIONS
- 12 FURTHER READING
WHAT IS IT?
Gaucher disease is a lysosomal storage disorder that is inherited in an autosomal recessive manner. It is caused by a deficiency in the glucocerebrosidase (β-glucosidase) enzyme.
WHY IS IT A PROBLEM?
The deficiency in the glucocerebrosidase enzyme leads to the accumulation of glucocerebroside (sphingolipid) in the lysosomes of cells, which will ultimately lead to the accumulation of this sphingolipid in macrophage lineage. These cells can in turn impair blood flow in the bones (bone crises) leading to complications like avascular necrosis, as well as causing other complications. The pathophysiology of bone complications is just one example of issues caused by this condition, and more points are addressed in the figure below.
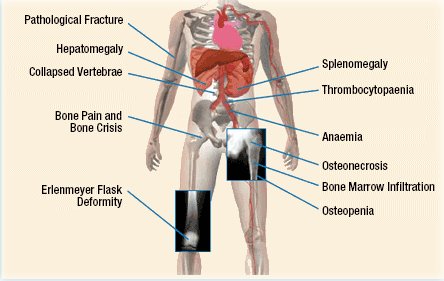
There are various subtypes for this condition each with a unique presentation (that we will not discuss here for the sake of simplicity).
WHAT MAKES US SUSPECT IT?
Risk factors:
Bone pain: that occurs due to bone crises, fractures, avascular necrosis. Commonly the pain will occur in the femur.
Hepatosplenomegaly that can be observed on physical exam.
HOW DO WE CONFIRM A DIAGNOSIS?
Pancytopenia (CBC) often times these patients will be deficient in white blood cells, erhytrocytes, and platelets
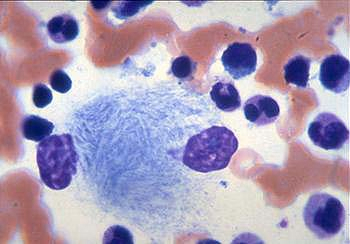
Bone marrow Gaucher cells: lipid filled macrophages that look akin to crumbled tissue paper. While not diagnostic on its own, this allows us to further explore this possible diagnosis.
Enzyme activity assay: diminished activity of glucosylceramidase in peripheral nucleated blood cells (such as leukocytes) will strongly suggest a diagnosis of Gaucher disease.
Sequencing: ultimately sequencing for the causal mutations will give a definitive diagnosis of Gaucher disease
HOW DO WE RULE OTHER DIAGNOSIS OUT?
Following the diagnostic path above should definitively rule out the possibility of other lysosomal storage diseases (whose enzyme activities can also be assayed). Sequencing other likely genes that can cause storage diseases may also be useful in trying to definitively rule out other conditions.
HOW DO WE TREAT IT?
Recombinant glucocerebrosidase: replacing the deficient enzyme corrects this disease ( alglucerase, Ceredase)
Substrate reduction therapy: If an individual cannot take enzyme replacement as a therapy (such as a pregnant woman) then medication that reduces the amount of sphingolipid (such as miglustat/Zavesca) can be used.
HOW WELL DO THE PATIENTS DO?
A normal life span is possible with Gaucher disease if it is properly managed.
WAS THERE A WAY TO PREVENT IT?
While the condition itself is not preventable, genetic counseling for couples who each have a autosomal recessive mutation can be informative with regards to communicating the risk of their children having this condition.
WHAT ELSE ARE WE WORRIED ABOUT?
Osteoporosis and subsequent fracture is common in this disease.
Avascular necrosis of femur is common due to impaired blood flow
OTHER HY FACTS?
Most common lysosomal storage disorder
Mutation on chromosome 1q21 (~200 different mutations are possible)
ARCHIVE OF STANDARDIZED EXAM QUESTIONS
This archive compiles standardized exam questions that relate to this topic.
FURTHER READING
Page Updated:01.21.2016